7 Figure 6
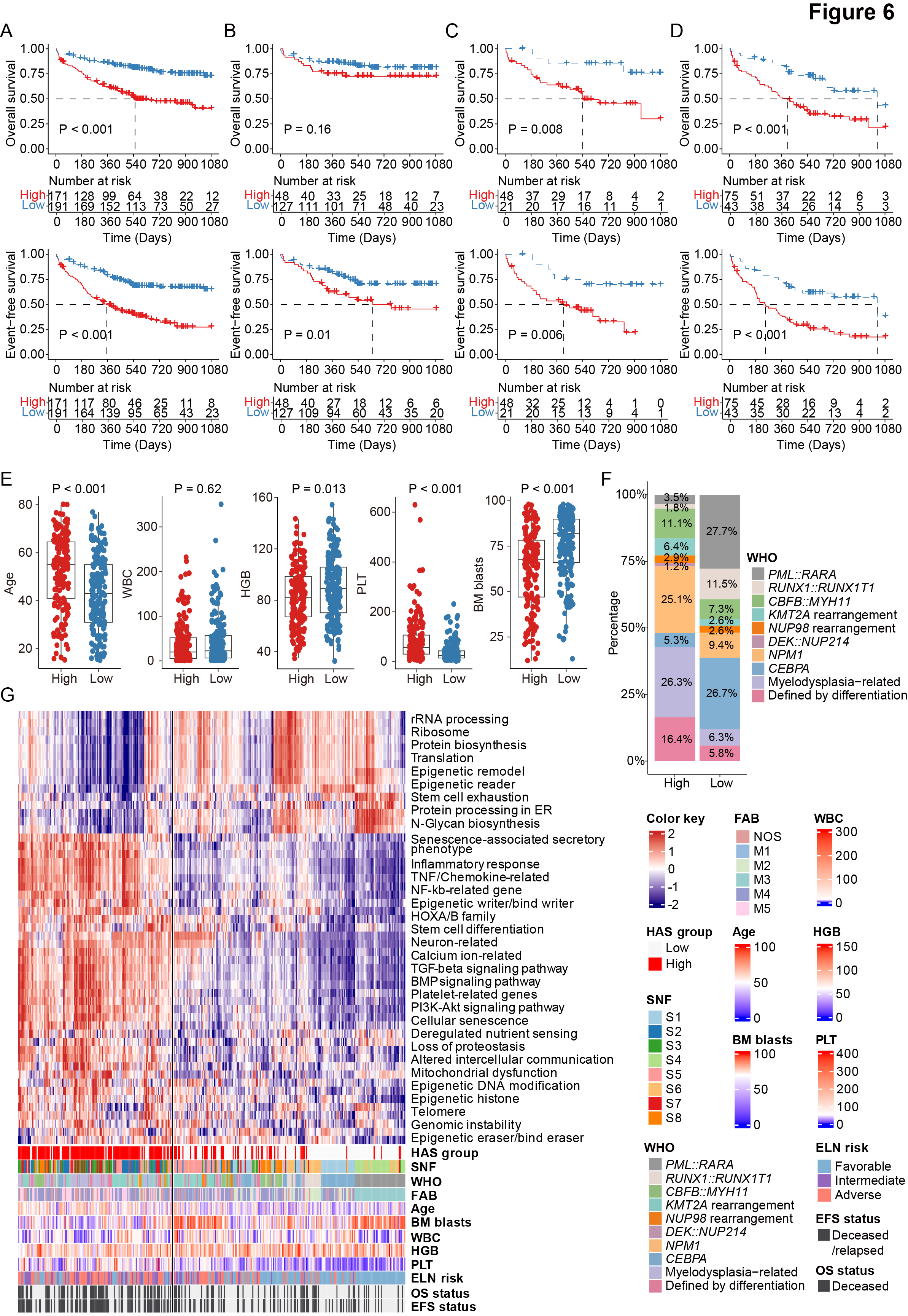
Figure 6. Clinical and molecular features of AML patients with high hematopoietic aging score.
A-D. Kaplan-Meier curves for overall survival (upper panels) and event-free survival (lower panels) in all AML patients (A) and patients with favorable (B), intermediate (C), and adverse (D) ELN risk stratified by the hematopoietic aging score (HAS), with the most obvious prognostic discrimination used as the cutoff value.
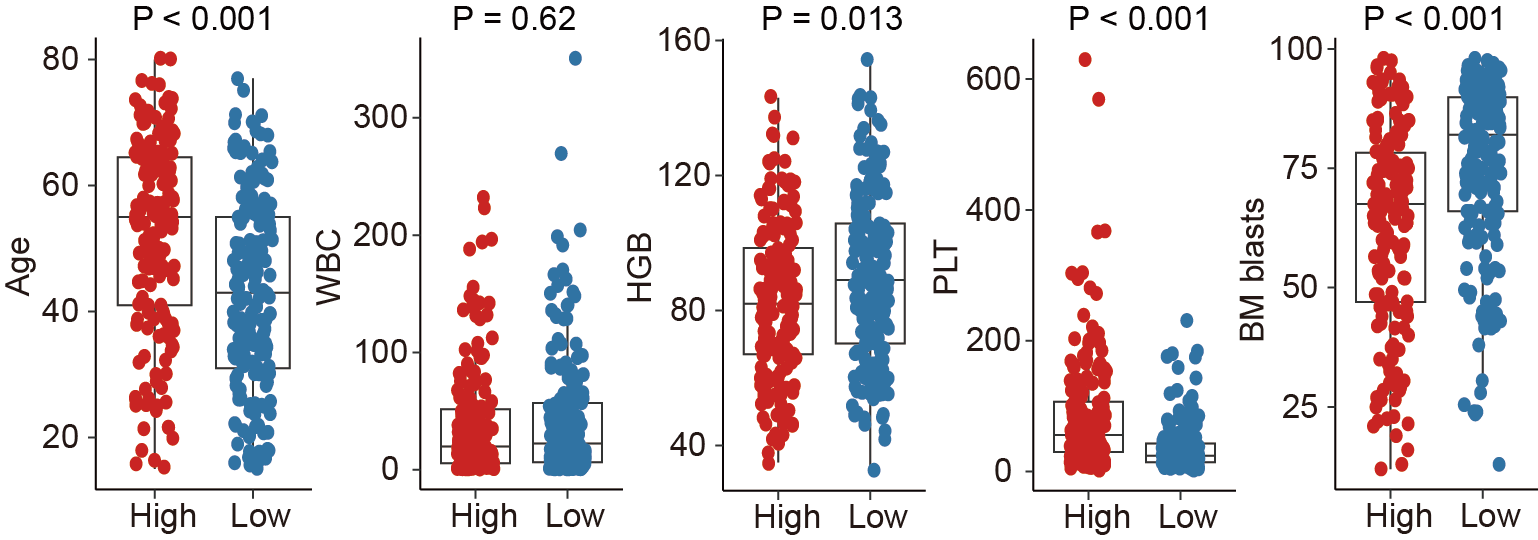
E. Comparison of age, WBC, HGB, PLT count, and BM blasts at diagnosis between high and low HAS groups.
F. Comparison of the proportion of WHO-defined entities between high and low HAS groups.
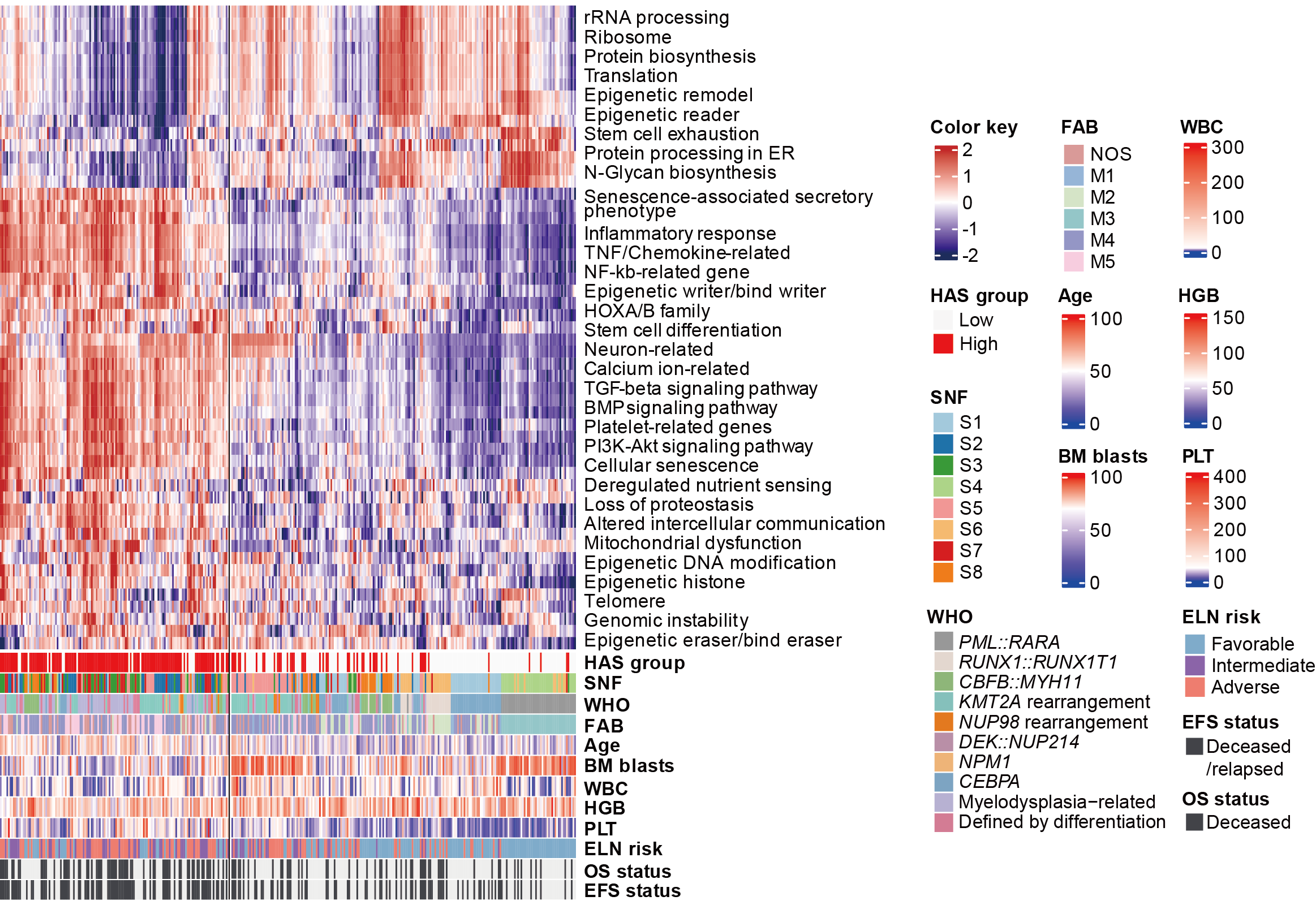
G. Heatmap demonstrating the enrichment of known aging-associated pathways in AML, including aging hallmarks, epigenetic gene sets, and our previously reported age-related enriched pathways. The HAS group and clinical features of patients are annotated below the heatmap. Each column represents a patient.
7.1 (A) Kaplan-Meier curves
Kaplan-Meier curves for overall survival (upper panels) and event-free survival (lower panels) in all AML patients with favorable
rm(list=ls())
library(readxl)
library(survminer)
library(survival)
score <- as.data.frame(read_xlsx("Input/Dataset S9 - hematopoietic lineage scores.xlsx", skip = 1))
data <- as.data.frame(read_xlsx("Input/Dataset S1 - clinical information.xlsx", skip = 1))
# 合并数据
data <- merge(data, score[, c('Sample_ID', 'HAS')], by = 'Sample_ID', all.x = TRUE, all.y = FALSE)
data <- subset(data, HAS != 0)
res.cut <- surv_cutpoint(data,
time ="OS",
event = "OS_status",
variables = "HAS",
minprop = 0.1)
data$HAS_group <- ifelse(data$HAS > res.cut$cutpoint$cutpoint,"High","Low")
data$HAS_group <- factor(data$HAS_group, levels = c("High","Low"))
data$OS[data$OS >= 1080] <- 1080
data$OS_status[data$OS >= 1080] <- 0
data$EFS[data$EFS >= 1080] <- 1080
data$EFS_status[data$EFS >= 1080] <- 0
fit <- survfit(Surv(OS,OS_status) ~ HAS_group, data)
ggsurvplot(fit, data = data,
linetype = "strata",
pval = TRUE,
risk.table = T, tables.height = 0.3,
#tables.theme = theme_cleantable(),
xlim = c(0,1080),
break.x.by = 180,
ylab="Overall survival",xlab = "Time (Days)",
surv.median.line = "hv",
font.x = c(14, "bold", "black"),
font.y = c(14, "bold", "black"),
font.tickslab = c(12, "plain", "black"),
palette = c("#E41A1C","#377EB8"),
legend.labs =c("High","Low"))
fit <- survfit(Surv(EFS,EFS_status) ~ HAS_group, data)
ggsurvplot(fit, data = data,
linetype = "strata",
pval = TRUE,
risk.table = T, tables.height = 0.3,
#tables.theme = theme_cleantable(),
xlim = c(0,1080),
break.x.by = 180,
ylab="Event-free survival",xlab = "Time (Days)",
surv.median.line = "hv",
font.x = c(14, "bold", "black"),
font.y = c(14, "bold", "black"),
font.tickslab = c(12, "plain", "black"),
palette = c("#E41A1C","#377EB8"),
legend.labs =c("High","Low"))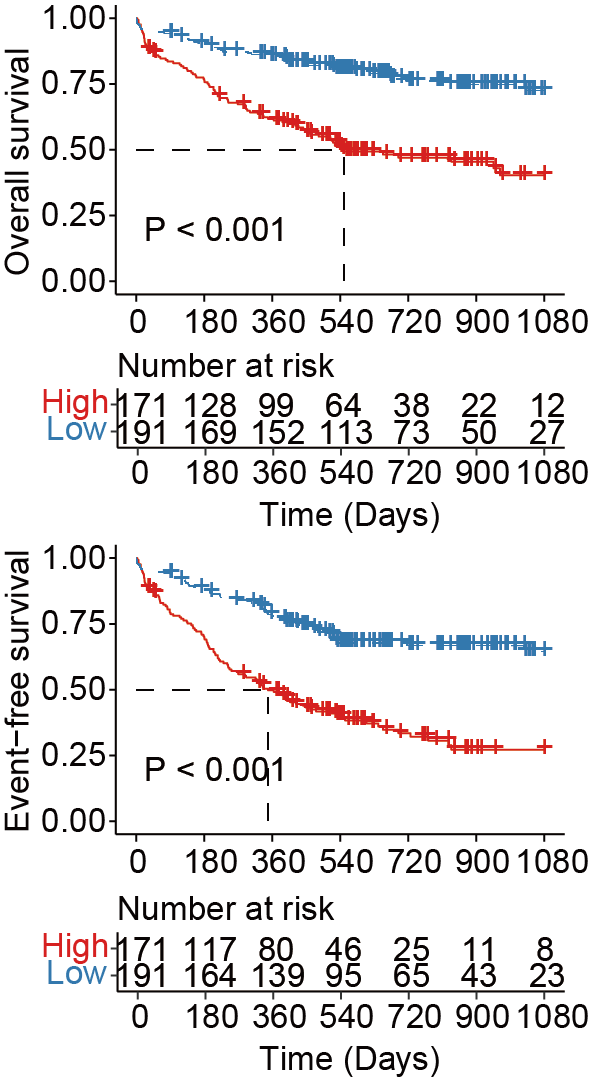
7.2 (E) Comparison
Comparison of age, WBC, HGB, PLT count, and BM blasts at diagnosis between high and low HAS groups.
rm(list = ls())
covariates <- c("Age", "WBC", "HGB", "PLT", "BM_blasts")
score <- as.data.frame(read_xlsx("Input/Dataset S9 - hematopoietic lineage scores.xlsx", skip = 1))
data <- as.data.frame(read_xlsx("Input/Dataset S1 - clinical information.xlsx", skip = 1))
# 合并数据
data <- merge(data, score[, c('Sample_ID', 'HAS')], by = 'Sample_ID', all.x = TRUE, all.y = FALSE)
data <- subset(data, HAS != 0)
res.cut <- surv_cutpoint(data,
time ="OS",
event = "OS_status",
variables = "HAS",
minprop = 0.1)
data$HAS_group <- ifelse(data$HAS > res.cut$cutpoint$cutpoint,"High","Low")
data$HAS_group <- factor(data$HAS_group, levels = c("High","Low"))
data$OS[data$OS >= 1080] <- 1080
data$OS_status[data$OS >= 1080] <- 0
data$EFS[data$EFS >= 1080] <- 1080
data$EFS_status[data$EFS >= 1080] <- 0
for (i in 1:(length(covariates))){
print(i)
p <- ggplot(data, aes(x = HAS_group, y = data[,covariates[i]])) +
geom_boxplot(outlier.shape = NA) +
geom_jitter(shape=21,na.rm = T,aes(fill=HAS_group,color=HAS_group),position = position_jitter(width = 0.2))+
xlab("Hematopoietic aging score")+ylab(covariates[i])+
scale_fill_manual(values = c("#E41A1C", "#377EB8"))+
scale_color_manual(values = c("#E41A1C", "#377EB8"))+
stat_compare_means(label = "p.format", method = "wilcox.test", hide.ns = F)+
scale_x_discrete(labels = c("High","Low"))+
theme_bw()+theme(legend.position = "none")+
theme(panel.grid.major=element_blank(),
panel.grid.minor=element_blank(),
axis.line = element_line(color = "black"),
panel.border = element_blank())
pdf(paste0(covariates[i], "_boxplot.pdf"),width = 3, height = 5)
print(p, newpage = FALSE)
dev.off()
}
7.3 (G) Heatmap with annotation
Heatmap demonstrating the enrichment of known aging-associated pathways in AML.
rm(list = ls())
library(ggpubr)
library(ggplot2)
library(cowplot)
library(ggthemes)
library(GSVA)
library(patchwork)
library(readxl)
library(export)
library(stringr)
library(openxlsx)
library(ComplexHeatmap)
library(circlize)
library(dplyr)
#step1--exp_and_geneset-------------------------------------------------------------
load("Input/pathway_heatmap.Rdata")
params <- list()
Age_color = colorRamp2(c(0, 50, 100), c("blue", "white", "red"))
WBC_color = colorRamp2(c(0, 10, 300), c("blue", "white", "red"))
HGB_color = colorRamp2(c(0, 60, 150), c("blue", "white", "red"))
PLT_color = colorRamp2(c(0, 50, 400), c("blue", "white", "red"))
BM_color = colorRamp2(c(0, 70, 100), c("blue", "white", "red"))
params$col <- list(
HAS_group = c("1" ="#ff0000", "0" = "#FAFAFA"),
SNF = c(SNF1="#A6CEE3", SNF2="#1F78B4", SNF3="#33A02C", SNF4="#B2DF8A" , SNF5="#FB9A99", SNF6="#FDBF6F", SNF7="#E31A1C", SNF8 ="#FF7F00"),
WHO = c(
"PML::RARA" = "#999999",
"RUNX1::RUNX1T1" = "#e7dad2",
"CBFB::MYH11" = "#96c37d",
"KMT2A rearrangement" = "#8ecfc9",
"NUP98 rearrangement" = "#FF7F00",
"DEK::NUP214" = "#c497b2",
"NPM1" = "#ffbe7a",
"CEBPA" = "#82b0d2",
"Myelodysplasia-related" = "#beb8dc",
"defined by differentiation" = "#e984a2"
),
Diagnosis = c(AML = "#DF9E9B", M1="#99BADF", M2 = "#D8E7CA", M3 = "#99CDCE", M4 = "#999ACD", M5 = "#FFD0E9"),
Age =Age_color,
BM_blasts =BM_color,
WBC = WBC_color,
HGB = HGB_color,
PLT = PLT_color,
ELN_risk = c("1" = "#80b1d3", "2" = "#9467bd", "3" ="#fb8072"),
OS_status = c("1" = "#434348", "0" = "#eeeeee"),
EFS_status = c("1" = "#434348", "0" = "#eeeeee"),
`PML::RARA` = c("1" = "#92C5DE", "0" = "#F7F7F7"),
`CBFB::MYH11` = c("1" = "#92C5DE", "0" = "#F7F7F7"),
`RUNX1::RUNX1T1` = c("1" = "#92C5DE", "0" = "#F7F7F7"),
`KMT2A fusions` = c("1" = "#92C5DE", "0" = "#F7F7F7"),
`NUP98 fusions` = c("1" = "#92C5DE", "0" = "#F7F7F7"),
`BCR::ABL1` = c("1" = "#92C5DE", "0" = "#F7F7F7"),
`Other fusions` = c("1" = "#92C5DE", "0" = "#F7F7F7"),
`Fusion genes` = c("1" = "#92C5DE", "0" = "#F7F7F7"),
Transcription.Factors = c("1" = "#92C5DE", "0" ="#F7F7F7" ),
Spliceosome = c("1" = "#92C5DE", "0" = "#F7F7F7"),
Tumor.Suppressors = c("1" = "#92C5DE", "0" = "#F7F7F7"),
NPM1 = c("1" = "#92C5DE", "0" = "#F7F7F7"),
DNA.Methylation = c("1" = "#92C5DE", "0" = "#F7F7F7"),
Activated.Signaling = c("1" = "#92C5DE", "0" = "#F7F7F7"),
Chromatin.Modifiers = c("1" = "#92C5DE", "0" = "#F7F7F7"),
Cohesin.Complex = c("1" = "#92C5DE", "0" = "#F7F7F7")
)
#加入数据信息:
params$HAS_group<-samples_heat$HAS_group
params$SNF <-samples_heat$SNF
params$WHO <-samples_heat$WHO
params$Diagnosis <-samples_heat$Diagnosis
params$Age <-samples_heat$Age
params$BM_blasts<-samples_heat$BM_blasts
params$WBC <- samples_heat$WBC
params$HGB <- samples_heat$HGB
params$PLT <- samples_heat$PLT
params$ELN_risk <- samples_heat$ELN_risk
params$OS_status <- samples_heat$OS_status
params$EFS_status <- samples_heat$EFS_status
params$`PML::RARA` <- samples_heat$`PML::RARA`
params$`CBFB::MYH11` <- samples_heat$`CBFB::MYH11`
params$`RUNX1::RUNX1T1` <- samples_heat$`RUNX1::RUNX1T1`
params$`KMT2A fusions` <- samples_heat$`KMT2A fusions`
params$`NUP98 fusions` <- samples_heat$`NUP98 fusions`
params$`BCR::ABL1` <- samples_heat$`BCR::ABL1`
params$`Other fusions` <- samples_heat$`Other fusions`
params$`Fusion genes` <- samples_heat$`Fusion genes`
params$Transcription.Factors <- samples_heat$Transcription.Factors
params$Spliceosome <- samples_heat$Spliceosome
params$Tumor.Suppressors <- samples_heat$Tumor.Suppressors
params$NPM1 <- samples_heat$NPM1
params$DNA.Methylation <- samples_heat$DNA.Methylation
params$Activated.Signaling <- samples_heat$Activated.Signaling
params$Chromatin.Modifiers <- samples_heat$Chromatin.Modifiers
params$Cohesin.Complex <-samples_heat$Cohesin.Complex
params$na_col <- "#FFFFFF"
#params$col$age <- col_fun_cont(samples_heat$age)
column_ha <- HeatmapAnnotation(
HAS_group = samples_heat$HAS_group,
SNF = samples_heat$SNF,
WHO = samples_heat$WHO,
Diagnosis = samples_heat$Diagnosis,
Age = samples_heat$Age,
BM_blasts = samples_heat$BM_blasts,
WBC = samples_heat$WBC,
HGB = samples_heat$HGB,
PLT = samples_heat$PLT,
ELN_risk = samples_heat$ELN_risk,
OS_status = samples_heat$OS_status,
EFS_status = samples_heat$EFS_status,
`PML::RARA` = samples_heat$`PML::RARA`,
`CBFB::MYH11` = samples_heat$`CBFB::MYH11`,
`RUNX1::RUNX1T1` = samples_heat$`RUNX1::RUNX1T1`,
`KMT2A fusions` = samples_heat$`KMT2A fusions`,
`NUP98 fusions` = samples_heat$`NUP98 fusions`,
`BCR::ABL1` = samples_heat$`BCR::ABL1`,
`Other fusions` = samples_heat$`Other fusions`,
`Fusion genes` = samples_heat$`Fusion genes`,
Transcription.Factors = samples_heat$Transcription.Factors,
Spliceosome = samples_heat$Spliceosome,
Tumor.Suppressors = samples_heat$Tumor.Suppressors,
NPM1 = samples_heat$NPM1,
DNA.Methylation = samples_heat$DNA.Methylation,
Activated.Signaling = samples_heat$Activated.Signaling,
Chromatin.Modifiers = samples_heat$Chromatin.Modifiers,
Cohesin.Complex = samples_heat$Cohesin.Complex,
col = params$col, # 确保颜色配置独立传递
na_col = "#FFFFFF"
)
set.seed(123)
p <- ComplexHeatmap::Heatmap(x_scale, col=col1,
column_names_gp = gpar(fontsize = 1),
clustering_distance_rows = function(x) as.dist((1-cor(t(x)))/2),
clustering_distance_columns = function(x) as.dist((1-cor(t(x)))/2),
clustering_method_rows = "ward.D",
clustering_method_columns = "ward.D",
cluster_columns = TRUE,
#column_split = samples_heat$age_cut,
bottom_annotation = column_ha,
use_raster = FALSE,
show_row_dend = FALSE,
show_column_dend = FALSE,
column_km = 2)
p<-draw(p)