6 Figure 5
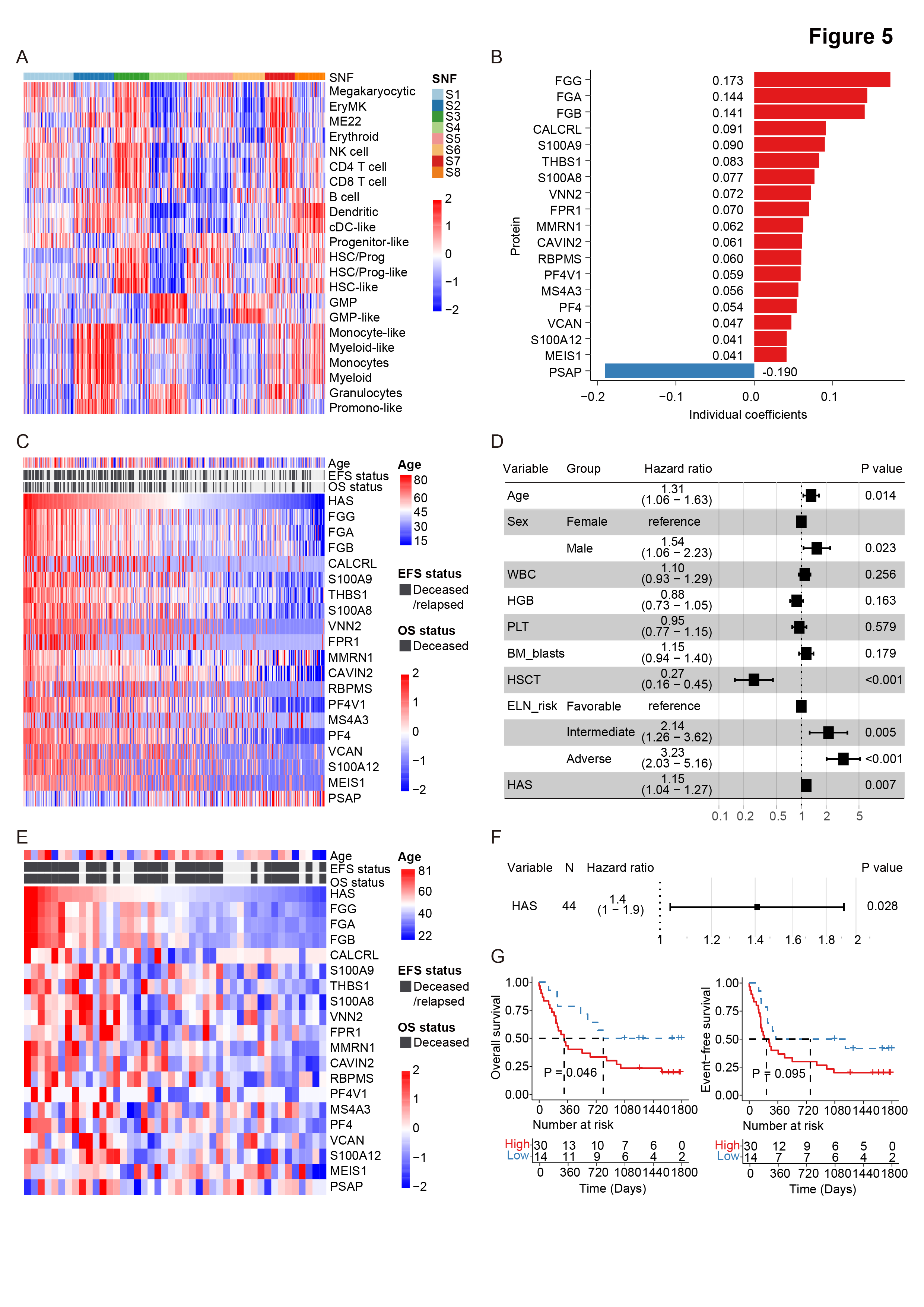
Figure 5. Development and validation of the hematopoietic aging score.
Heatmap depicting the protein abundance of various hematopoietic lineage markers across the eight proteomic subtypes.
Proteins included in the hematopoietic aging score (HAS). The final score is calculated as the weighted sum of abundance of the 19 proteins. Proteins in red and blue are predictive for a poor and good prognosis, respectively. Coefficients of these proteins in univariate Cox regression analysis are denoted.
Heatmap showing the established HAS and the abundance of the 19 proteins, with patients arranged according to the magnitude of their scores. Age and prognosis of patients are annotated above the heatmap.
Forest plots of multivariate Cox regression analysis for overall survival in AML patients. Hazard ratios (HRs) and 95% confidence intervals (CIs) are listed next to variables. The dotted vertical line indicates an HR of 1. Black boxes denote the HR of each variable, and horizontal lines show 95% CIs. P values indicating the significance are shown on the right.
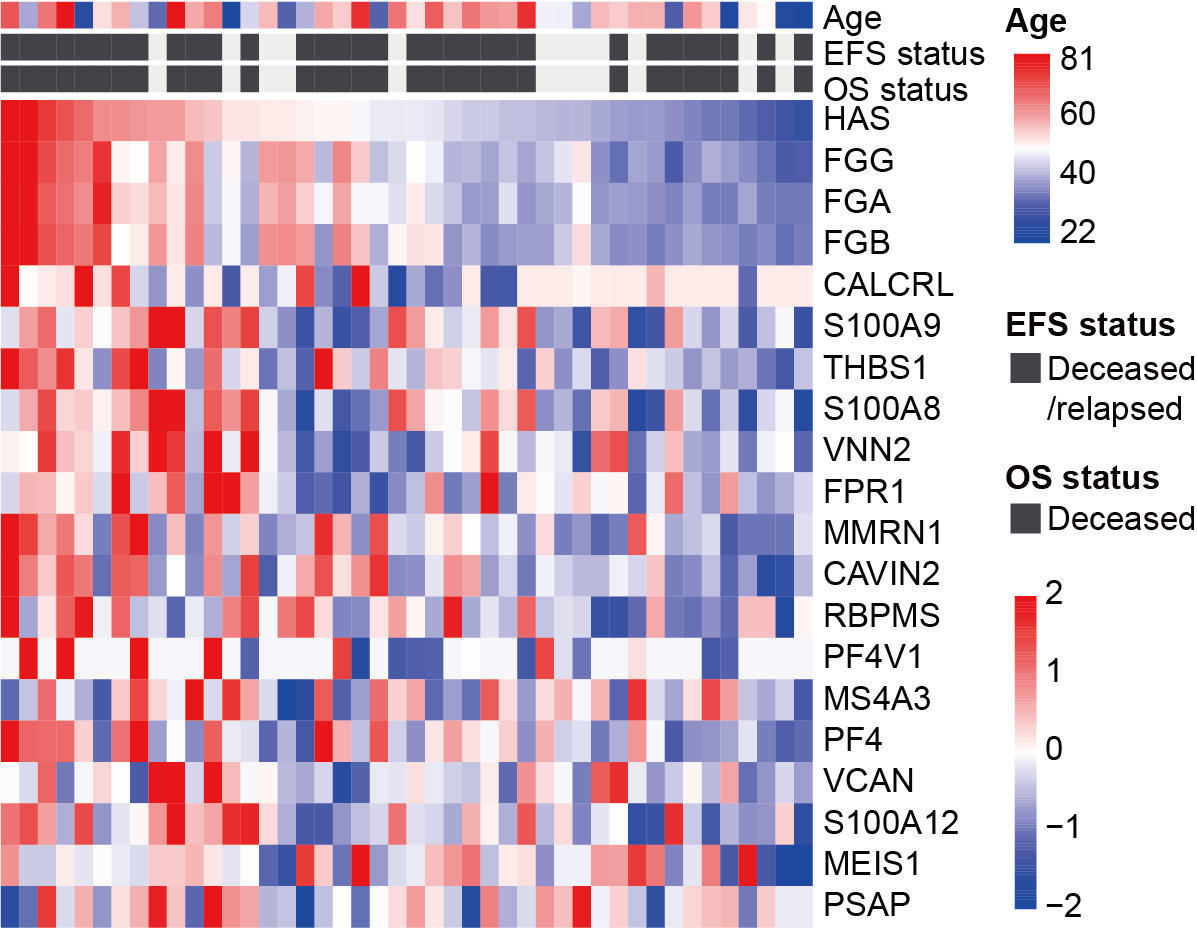
Heatmap of markers incorporated into the HAS in an independent external dataset.
The prognostic value of the HAS in univariate Cox regression in an independent external dataset.
Kaplan-Meier curves for overall survival (left panel) and event-free survival (right panel) stratified by the HAS in external dataset, with the most obvious prognostic discrimination used as the cutoff value.
6.1 (A) Various hematopoietic lineage scores in proteomic subtypes
Heatmap depicting the levels of various hematopoietic lineage scores in proteomic subtypes.
rm(list = ls())
#-----------------------------------------------------------------------------
#Step 1: Load data
#-----------------------------------------------------------------------------
AML_data<-readRDS("Input/AML_data.rds")
AML_sampleinfo_20240214<-AML_data$sampleinfo
AML_sampleinfo_20240214 <- AML_sampleinfo_20240214[,c('DIA_ID', 'OS', 'OS_status', 'HSCT', 'Sex', 'WBC', 'ELN_risk_2017', 'EFS_status')]
clinical.features <- AML_data$clinical_features
clinical.features <- clinical.features[clinical.features$SNF_subtype!="NA",]
protein.abundance<-AML_data$CorrectProteomics
protein.meta <- protein.abundance[, c(1,2)]
rownames(protein.abundance) <- protein.abundance[,1]
protein.abundance <- protein.abundance[, -c(1,2)]
protein.abundance <- t(protein.abundance)
protein.abundance <- protein.abundance[which(rownames(protein.abundance) %in% clinical.features$Sample_ID),]
#-----------------------------------------------------------------------------
#Step 2: Marker proteins
#-----------------------------------------------------------------------------
gs <- readRDS("Input/fig5files/gs.RDS")
Supplementary_41590_2017_BFni3693_MOESM12_ESM <- read.table('Input/fig5files/41590_2017_BFni3693_MOESM12_ESM.txt', sep='\t', header = TRUE)
BCell.NI <- Supplementary_41590_2017_BFni3693_MOESM12_ESM$gene.names[which(Supplementary_41590_2017_BFni3693_MOESM12_ESM$BCell.vs.Rest == '+')]
BCell.NI <- strsplit(BCell.NI, ";") %>% unlist() %>% toupper()
CD4TCell.NI <- Supplementary_41590_2017_BFni3693_MOESM12_ESM$gene.names[which(Supplementary_41590_2017_BFni3693_MOESM12_ESM$CD4TCell.vs.Rest == '+')]
CD4TCell.NI <- strsplit(CD4TCell.NI, ";") %>% unlist() %>% toupper()
CD8TCell.NI <- Supplementary_41590_2017_BFni3693_MOESM12_ESM$gene.names[which(Supplementary_41590_2017_BFni3693_MOESM12_ESM$CD8TCell.vs.Rest == '+')]
CD8TCell.NI <- strsplit(CD8TCell.NI, ";") %>% unlist() %>% toupper()
Dendritic.NI <- Supplementary_41590_2017_BFni3693_MOESM12_ESM$gene.names[which(Supplementary_41590_2017_BFni3693_MOESM12_ESM$Dendritic.vs.Rest == '+')]
Dendritic.NI <- strsplit(Dendritic.NI, ";") %>% unlist() %>% toupper()
Granulocytes.NI <- Supplementary_41590_2017_BFni3693_MOESM12_ESM$gene.names[which(Supplementary_41590_2017_BFni3693_MOESM12_ESM$Granulocytes.vs.Rest == '+')]
Granulocytes.NI <- strsplit(Granulocytes.NI, ";") %>% unlist() %>% toupper()
Monocytes.NI <- Supplementary_41590_2017_BFni3693_MOESM12_ESM$gene.names[which(Supplementary_41590_2017_BFni3693_MOESM12_ESM$Monocytes.vs.Rest == '+')]
Monocytes.NI <- strsplit(Monocytes.NI, ";") %>% unlist() %>% toupper()
NKCell.NI <- Supplementary_41590_2017_BFni3693_MOESM12_ESM$gene.names[which(Supplementary_41590_2017_BFni3693_MOESM12_ESM$NKCell.vs.Rest == '+')]
NKCell.NI <- strsplit(NKCell.NI, ";") %>% unlist() %>% toupper()
EryMK <- toupper(c(
# 红系
'CD71','TFRC','CD36','CD235a','GYPA',
# 巨核
'CD41','ITGA2B','CD61','ITGB3','CD42b','GP1BA','GP1BB',
# 红系
'GATA1', 'CA1', 'HBA1', 'HBB', 'ALAD',
# 巨核
'GATA2', 'ZFPM1', 'FLI1', 'NFE2', 'PF4'))
marker.genes <- unlist(strsplit(Supplementary_41590_2017_BFni3693_MOESM12_ESM$gene.names, ";"))
marker.genes <- unique(c(marker.genes, EryMK))
marker.genes <- toupper(marker.genes)
markers.1_s2.0_S0092867419300947_mmc3 <- read.table('Input/fig5files/1-s2.0-S0092867419300947-mmc3.txt', sep='\t', header=TRUE)
markers.1_s2.0_S0092867419300947_mmc3 <- markers.1_s2.0_S0092867419300947_mmc3[,-12]
protein.meta$PG.Genes <- toupper(protein.meta$PG.Genes)
#-----------------------------------------------------------------------------
#Step 3: Transfer ID and symbols
#-----------------------------------------------------------------------------
protein.meta$PG.Genes <- toupper(protein.meta$PG.Genes)
library(tidyr)
protein.meta_expanded <- protein.meta %>%
separate_rows(PG.Genes, sep = ";")
protein.meta_expanded <- as.data.frame(protein.meta_expanded)
r_list <- list(ATHL1="PGGHG", CXorf57 = "RADX", EMR1 = "ADGRE1", EMR2 = "ADGRE2", EMR3 = "ADGRE3",
FAIM3 = "FCMR", FAM198B = "GASK1B", FAM212B= "RIPOR2", FAM212B = "INKA2",
FAM65B = "RIPOR2", FLJ13197 = "KLF3-AS1", GPR1 = "CMKLR2", GPR97= "ADGRG3",
HIST1H2AE = "H2AC8", HIST1H2BG = "H2BC8", KIAA0226L = "RUBCNL",
KIAA0754 = "MACF1", KIRREL = "KIRREL1", LRMP = "IRAG2", "MARCH3" = "MARCHF3",
"SEPT5" = "SEPTIN5", "SEPT8" = "SEPTIN8", VNN3 = "VNN3P")
ref_list <- list(GUCY1A1="GUCY1A3", TENT5A = "FAM46A", "H1-0"="H1F0", MACROH2A1 = "H2AFY", "SNHG32" = "C6orf48")
CD.markers <- read.table('Input/fig5files/CD.markers.txt', sep='\t',header=TRUE)
#-----------------------------------------------------------------------------
#Step 4: Calculate scores
#-----------------------------------------------------------------------------
names(gs) <- stringr::str_replace_all(names(gs), "-", ".")
names(gs) <- paste(names(gs),'.gs', sep='')
TableS5.Cellcyclerelatedgenes <- read.table('Input/fig5files/Table S5. Cell cycle related genes.txt')
TableS5.Cellcyclerelatedgenes <- toupper(TableS5.Cellcyclerelatedgenes[,1])
Markers.list <- list(
BCell.NI=toupper(BCell.NI),
CD4TCell.NI=toupper(CD4TCell.NI),
CD8TCell.NI=toupper(CD8TCell.NI),
Dendritic.NI=toupper(Dendritic.NI),
Granulocytes.NI=toupper(Granulocytes.NI),
Monocytes.NI=toupper(Monocytes.NI),
NKCell.NI=toupper(NKCell.NI),
EryMK=toupper(EryMK),
HSC_Prog.gs=toupper(gs[['HSC_Prog.gs']]),
GMP.gs=toupper(gs[['GMP.gs']]),
Myeloid.gs=toupper(gs[['Myeloid.gs']]),
HSC_Prog.like.gs=toupper(gs[['HSC_Prog.like.gs']]),
GMP_like.gs=toupper(gs[['GMP_like.gs']]),
Myeloid_like.gs=toupper(gs[['Myeloid_like.gs']]),
HSC.like.gs=toupper(gs[['HSC.like.gs']]),
Progenitor.like.gs=toupper(gs[['Progenitor.like.gs']]),
Promono.like.gs=toupper(gs[['Promono.like.gs']]),
Monocyte.like.gs=toupper(gs[['Monocyte.like.gs']]),
cDC.like.gs=toupper(gs[['cDC.like.gs']]),
Erythroid.gs=toupper(gs[['Erythroid.gs']]),
Megakaryocytic.gs=toupper(gs[['Megakaryocytic.gs']])
)
saveRDS(Markers.list, file='Input/fig5files/Markers.list.Rds')
Score.matrix <- as.data.frame(matrix(NA, nrow=nrow(protein.abundance), ncol=length(Markers.list)))
rownames(Score.matrix) <- rownames(protein.abundance)
colnames(Score.matrix) <- names(Markers.list)
for(j_col in 1:ncol(Score.matrix)){
Markers.list.temp <- Markers.list[[colnames(Score.matrix)[j_col]]]
Markers.list.temp.PG.ProteinGroups <- plyr::mapvalues(Markers.list.temp,
from=protein.meta_expanded$PG.Genes,
to=protein.meta_expanded$PG.ProteinGroups,
warn_missing=FALSE)
Score.matrix[,colnames(Score.matrix)[j_col]] <- rowMeans(protein.abundance[,colnames(protein.abundance)%in%Markers.list.temp.PG.ProteinGroups])
}
expression_df <- Score.matrix
annotation <- data.frame(Group = plyr::mapvalues(rownames(expression_df),
from=clinical.features$Sample_ID,
to=clinical.features$SNF_subtype))
rownames(annotation) <- rownames(expression_df)
annotation$samples <- rownames(annotation)
annotation$Group <- factor(annotation$Group, levels = c('SNF1','SNF2','SNF3','SNF4','SNF5','SNF6','SNF7','SNF8'))
annotation$OS_status <- factor(plyr::mapvalues(annotation$samples,
from=AML_sampleinfo_20240214$DIA_ID,
to=AML_sampleinfo_20240214$OS_status), levels=c(0,1))
annotation$EFS_status <- factor(plyr::mapvalues(annotation$samples,
from=AML_sampleinfo_20240214$DIA_ID,
to=AML_sampleinfo_20240214$EFS_status), levels=c(0,1))
annotation <- annotation[order(annotation$Group, decreasing = FALSE),]
expression_df <- expression_df[annotation$samples,]
expression_df_normalized <- scale(expression_df)
expression_df_normalized <- pmax(pmin(expression_df_normalized, 2), -2)
annotation <- annotation[,-2]
options(repr.plot.width=8, repr.plot.height=6)
my_palette <- colorRamp2(c(-2, 0, 2),
c("navy", "white","firebrick3"))
SNF_color <- c("#A6CEE3", "#1F78B4", "#33A02C", "#B2DF8A", "#FB9A99", "#FDBF6F", "#E31A1C", "#FF7F00")
names(SNF_color) <- c('SNF1','SNF2','SNF3','SNF4','SNF5','SNF6','SNF7','SNF8')
OS_color <- c("#eeeeee", "#434348")
names(OS_color) <- c('0','1')
EFS_color <- c("#eeeeee", "#434348")
names(EFS_color) <- c('0','1')
annotation_colors <- list(Group = SNF_color,
OS_status = OS_color,
EFS_status = EFS_color)
p<-pheatmap(t(expression_df_normalized),
annotation_col = annotation,
scale = "none",
show_rownames = TRUE,
show_colnames = FALSE,
cluster_rows = TRUE,
cluster_cols = FALSE,
color = my_palette,
annotation_colors = annotation_colors,
treeheight_row = 0,
width = 8,
height= 5,
file="Output/Figure5/heatmap.lineageScore.pdf")
draw(p)
save.image('Input/fig5files/lineageScores.RData')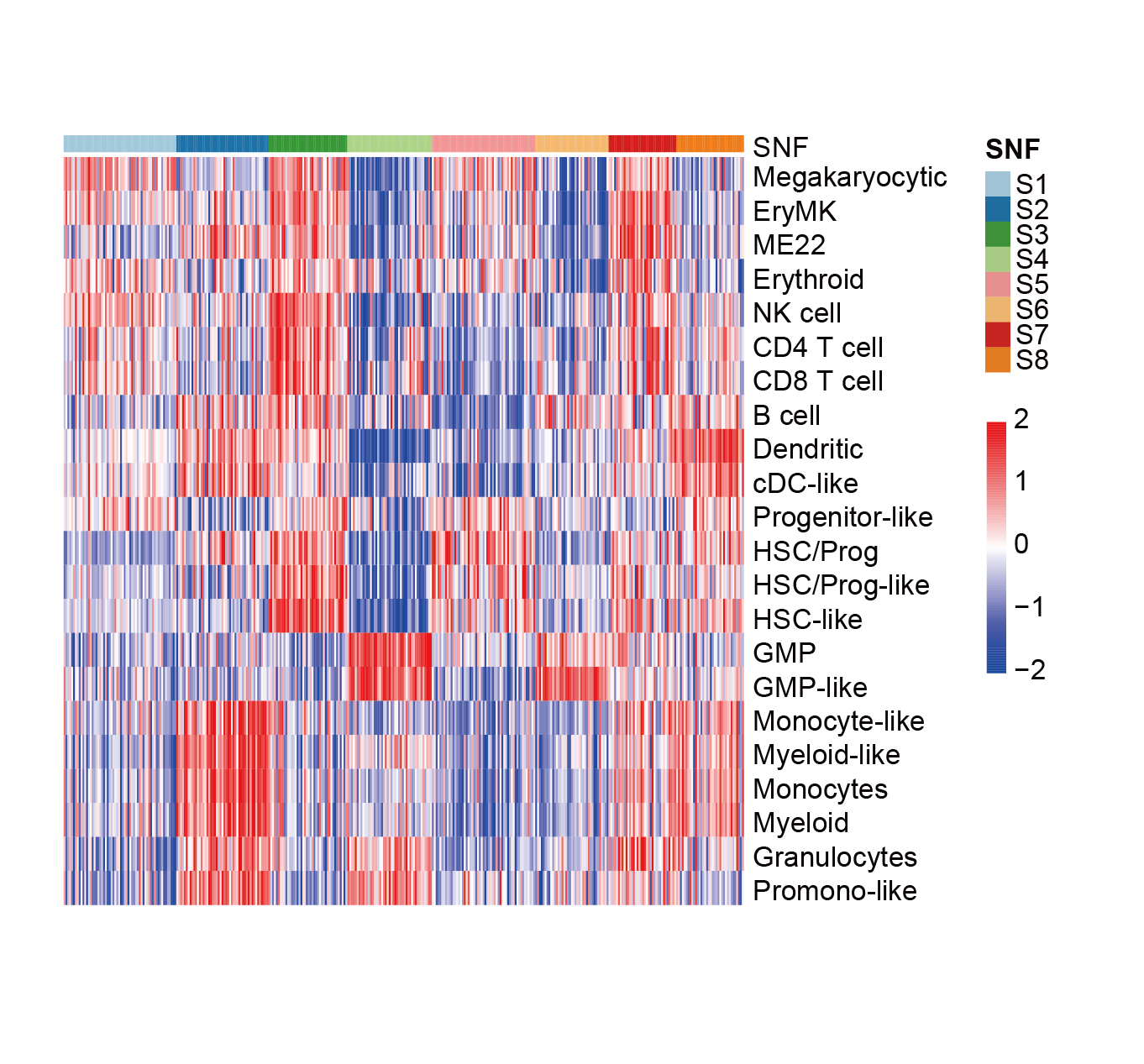
6.2 (B) Proteins included in the hematopoietic aging score (HAS).
The final score is calculated as the weighted sum of abundance of the 19 proteins. Proteins in red and blue are predictive for a poor and good prognosis, respectively. Coefficients of these proteins in univariate Cox regression analysis are denoted.
rm(list = ls())
coef <- as.data.frame(read.csv("Input/overlap.proteins.csv"))
coef$category <- ifelse(coef$coef > 0, "pos", "neg")
ggplot(coef, aes(x = reorder(Protein, coef), y = coef, fill = category)) +
geom_bar(stat = "identity") +
coord_flip() +
scale_fill_manual(values = c("pos" = "#E41A1C", "neg" = "#377EB8")) +
labs(x = "Protein", y = "Individual coefficients") +
theme_minimal() +
theme_bw()+theme(legend.position = "none")+
theme(panel.grid.major=element_blank(),
panel.grid.minor=element_blank(),
axis.line = element_line(color = "black"),
panel.border = element_blank())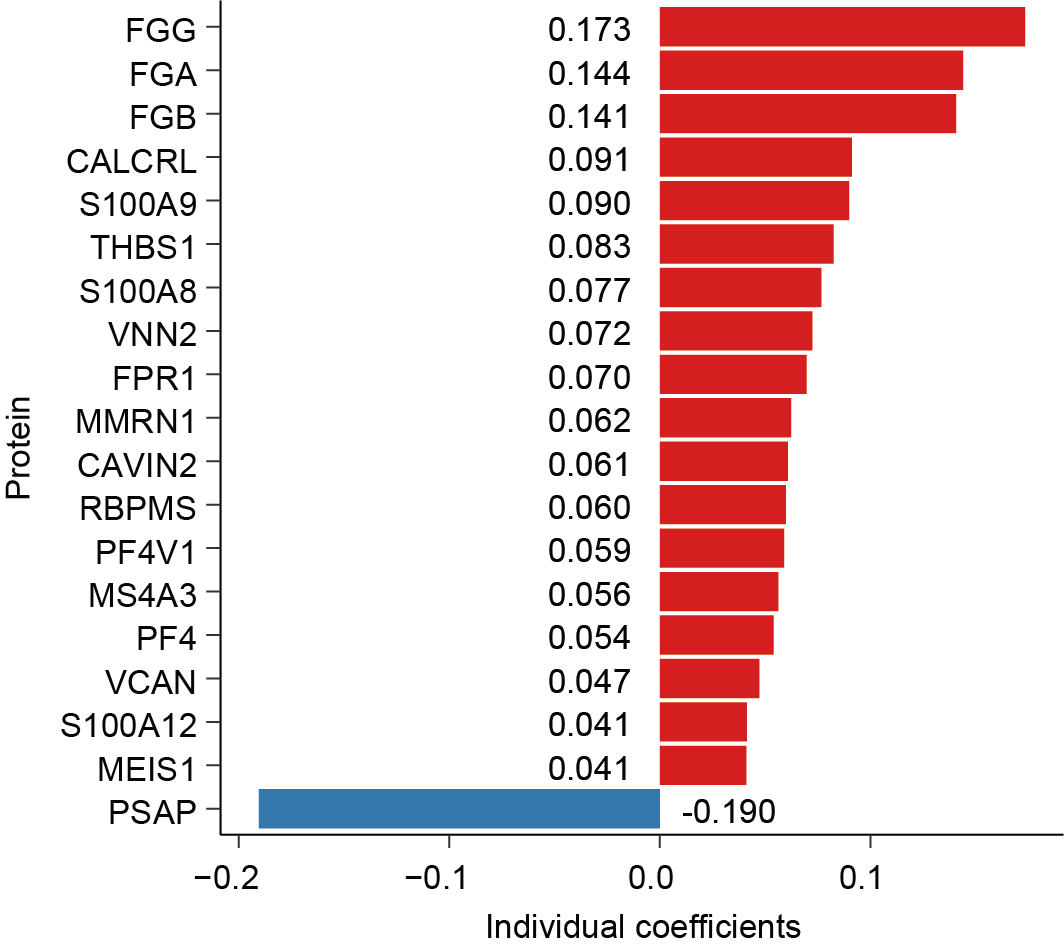
6.3 (C) Heatmap
Heatmap showing the established HAS and the abundance of the 19 proteins, with patients arranged according to the magnitude of their scores. Age and prognosis of patients are annotated above the heatmap.
rm(list=ls())
AML_data<-readRDS("Input/AML_data.rds")
AML_sampleinfo_20240214<-AML_data$sampleinfo
AML_sampleinfo_20240214 <- AML_sampleinfo_20240214[,c('DIA_ID', 'OS', 'OS_status', 'HSCT', 'Sex', 'WBC', 'ELN_risk_2017', 'EFS_status')]
clinical.features <- AML_data$clinical_features
clinical.features <- clinical.features[clinical.features$SNF_subtype!="NA",]
protein.abundance<-AML_data$CorrectProteomics
protein.meta <- protein.abundance[, c(1,2)]
rownames(protein.abundance) <- protein.abundance[,1]
protein.abundance <- protein.abundance[, -c(1,2)]
protein.abundance <- t(protein.abundance)
protein.abundance <- protein.abundance[which(rownames(protein.abundance) %in% clinical.features$Sample_ID),]
Supplementary_41590_2017_BFni3693_MOESM12_ESM <- read.table('Input/fig5files/41590_2017_BFni3693_MOESM12_ESM.txt', sep='\t', header = TRUE)
marker.genes <- unlist(strsplit(Supplementary_41590_2017_BFni3693_MOESM12_ESM$gene.names, ";"))
EryAndMega.markers <- toupper(c('CD71','TFRC','CD36','CD235a','GYPA','CD41','ITGA2B','CD61','ITGB3','CD42b','GP1BA','GP1BB'))
marker.genes <- unique(c(marker.genes, EryAndMega.markers))
marker.genes <- toupper(marker.genes)
protein.meta$PG.Genes <- toupper(protein.meta$PG.Genes)
library(tidyr)
protein.meta_expanded <- protein.meta %>%
separate_rows(PG.Genes, sep = ";")
protein.meta_expanded <- as.data.frame(protein.meta_expanded)
marker.genes <- c('PF4','ZNF16','NRIP1','CALCRL','S100A9','THBS1','S100A8','VNN2','FPR1','MMRN1','RBPMS','MS4A3','TAL1','VCAN','S100A12','MEIS1')
protein.meta_expanded <- protein.meta_expanded[which(protein.meta_expanded$PG.Genes %in% marker.genes), ]
library(pheatmap)
expression_df <- protein.abundance[,protein.meta_expanded$PG.ProteinGroups]
annotation <- data.frame(Group = plyr::mapvalues(rownames(expression_df),
from=clinical.features$Sample_ID,
to=clinical.features$SNF_subtype))
rownames(annotation) <- rownames(expression_df)
annotation$samples <- rownames(annotation)
annotation$Group <- factor(annotation$Group, levels = c('SNF1','SNF2','SNF3','SNF4','SNF5','SNF6','SNF7','SNF8'))
annotation$OS_status <- factor(plyr::mapvalues(annotation$samples,
from=AML_sampleinfo_20240214$DIA_ID,
to=AML_sampleinfo_20240214$OS_status), levels=c(0,1))
annotation$EFS_status <- factor(plyr::mapvalues(annotation$samples,
from=AML_sampleinfo_20240214$DIA_ID,
to=AML_sampleinfo_20240214$EFS_status), levels=c(0,1))
annotation <- annotation[order(annotation$Group, decreasing = FALSE),]
expression_df <- expression_df[annotation$samples,]
RJ.Score.matrix <- read.csv(file='Input/fig5files/RJ.Score.matrix.csv', header = TRUE, row.names = 1)
expression_df <- as.data.frame(expression_df)
expression_df$ZXSL.MK.other <- as.numeric(plyr::mapvalues(rownames(expression_df),
from=rownames(RJ.Score.matrix),
to=RJ.Score.matrix$ZXSL.MK.other))
expression_df <- expression_df[order(expression_df$ZXSL.MK.other, decreasing = TRUE),]
expression_df_normalized <- scale(expression_df)
expression_df_normalized[which(expression_df_normalized > 2)] <- 2
expression_df_normalized[which(expression_df_normalized < -2)] <- -2
colnames(expression_df_normalized) <- plyr::mapvalues(colnames(expression_df_normalized),
from=protein.meta_expanded$PG.ProteinGroups,
to=protein.meta_expanded$PG.Genes)
annotation <- annotation[,-2]
my_palette <- readRDS("Input/my_palette_1.rds")
SNF_color <- c("#A6CEE3", "#1F78B4", "#33A02C", "#B2DF8A", "#FB9A99", "#FDBF6F", "#E31A1C", "#FF7F00")
names(SNF_color) <- c('SNF1','SNF2','SNF3','SNF4','SNF5','SNF6','SNF7','SNF8')
OS_color <- c("#eeeeee", "#434348")
names(OS_color) <- c('0','1')
EFS_color <- c("#eeeeee", "#434348")
names(EFS_color) <- c('0','1')
annotation_colors <- list(Group = SNF_color,
OS_status = OS_color,
EFS_status = EFS_color)
plot <- pheatmap(t(expression_df_normalized),
annotation_col = annotation,
scale = "none",
show_rownames = TRUE,
show_colnames = FALSE,
cluster_rows = TRUE,
cluster_cols = FALSE,
color = my_palette,
annotation_colors = annotation_colors,
treeheight_row = 0,
width = 8,
height= 5)
rownames(t(expression_df_normalized))[plot$tree_row$order]
pheatmap(t(expression_df_normalized)[c('ZXSL.MK.other','PF4','ZNF16','MS4A3','VCAN','FPR1','VNN2','S100A12','S100A8','S100A9','CALCRL','MEIS1','RBPMS','TAL1','THBS1','NRIP1','MMRN1'),],
annotation_col = annotation,
scale = "none",
show_rownames = TRUE,
show_colnames = FALSE,
cluster_rows = FALSE,
cluster_cols = FALSE,
color = my_palette,
annotation_colors = annotation_colors,
treeheight_row = 0,
width = 8,
height= 5,
file="Output/Figure5/heatmap.select.markers.pdf")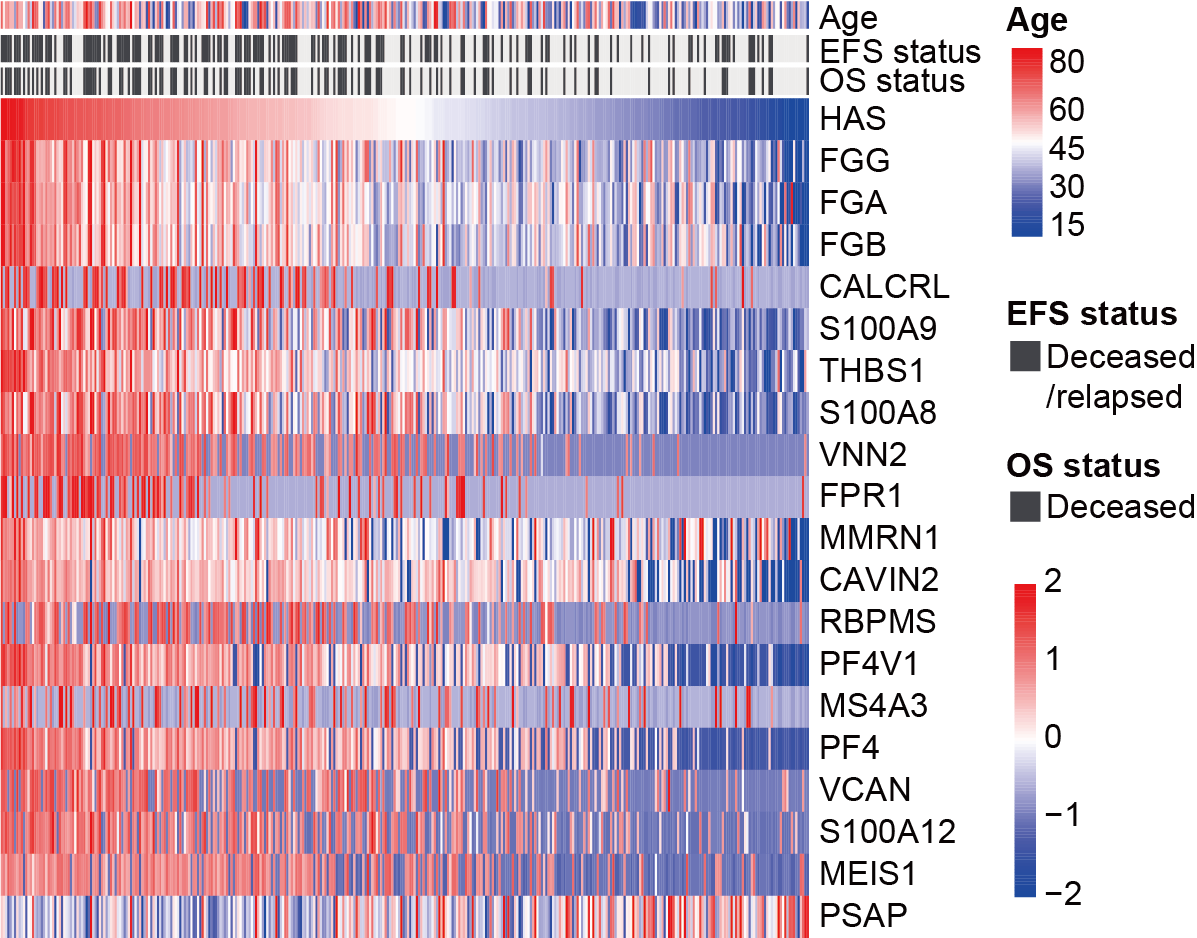
6.4 (D) Forest plots
Forest plots of multivariate Cox regression analysis for overall survival in AML patients. Hazard ratios (HRs) and 95% confidence intervals (CIs) are listed next to variables. The dotted vertical line indicates an HR of 1. Black boxes denote the HR of each variable, and horizontal lines show 95% CIs. P values indicating the significance are shown on the right.
rm(list=ls())
library(survival)
library(readxl)
library(survminer)
# 读取数据
score <- as.data.frame(read_xlsx("Input/Dataset S9 - hematopoietic lineage scores.xlsx", skip = 1))
data <- as.data.frame(read_xlsx("Input/Dataset S1 - clinical information.xlsx", skip = 1))
# 合并数据
data <- merge(data, score[, c('Sample_ID', 'HAS')], by = 'Sample_ID', all.x = TRUE, all.y = FALSE)
# 过滤HAS非零的样本
data <- subset(data, HAS != 0)
# 处理协变量
covariates <- c("Age", "WBC", "HGB", "PLT", "BM_blasts")
data[, covariates] <- lapply(data[, covariates], as.numeric)
# 标准化并确保结果为向量
data[, covariates] <- as.data.frame(scale(data[, covariates]))
# 删除缺失值(如有必要)
data <- na.omit(data)
# 拟合Cox模型并保存数据
fit <- coxph(
Surv(OS, OS_status) ~ Age + Sex + WBC + HGB + PLT + BM_blasts + HSCT + ELN_risk_2017 + HAS,
data = data,
model = TRUE # 确保模型保存数据
)
# 生成森林图
ggforest(fit, data = data) # 显式传递数据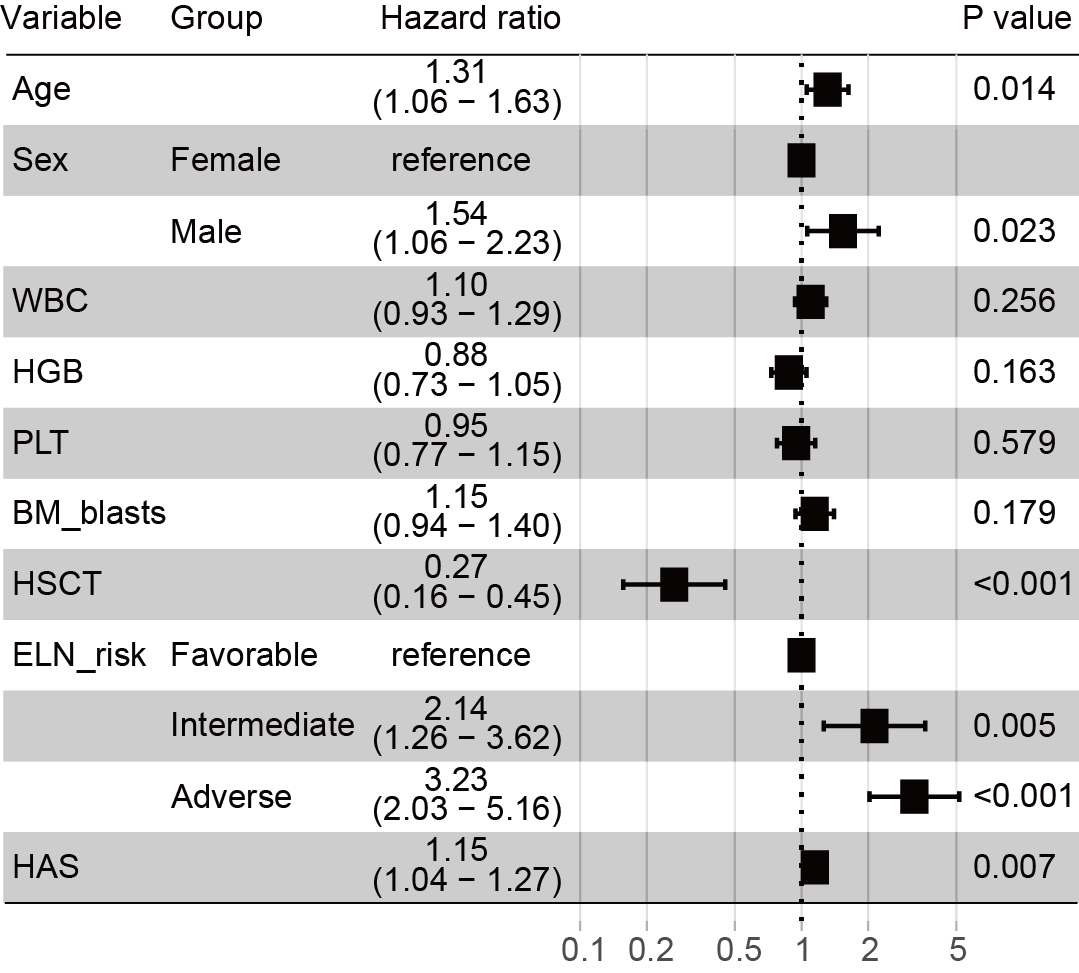
6.5 (E) Markers incorporated into the hematopoietic lineage scores.
Heatmap of markers incorporated into the hematopoietic lineage scores in an independent external dataset.
rm(list=ls())
load('Input/fig5files/lineageScores.RData')
Markers.list.combined <- readRDS(file='Input/fig5files/Markers.list.combined.Rds')
bloodBLD2022016033.suppl3 <- readRDS('Input/fig5files/bloodBLD2022016033.suppl3.Rds')
bloodbld2022016033.suppl4.TMT.matrix <- readRDS('Input/fig5files/bloodbld2022016033.suppl4.TMT.matrix.Rds')
protein.abundance <- bloodbld2022016033.suppl4.TMT.matrix
Score.matrix <- read.csv(file='Input/fig5files/Blood.Score.matrix.csv', row.names=1)
marker.genes <- c('PF4','ZNF16','NRIP1','CALCRL','S100A9','THBS1','S100A8','VNN2','FPR1','MMRN1','RBPMS','MS4A3','TAL1','VCAN','S100A12','MEIS1')
expression_df <- protein.abundance[,which(colnames(protein.abundance)%in%marker.genes)]
expression_df$ZXSL.MK.other <- as.numeric(plyr::mapvalues(rownames(expression_df), from=rownames(Score.matrix), to=Score.matrix$ZXSL.MK.other))
expression_df <- expression_df[, c('ZXSL.MK.other','PF4','ZNF16','MS4A3','VCAN','FPR1','VNN2','S100A12','S100A8','S100A9','CALCRL','MEIS1','RBPMS','TAL1','THBS1','NRIP1','MMRN1')]
annotation <- data.frame(OS_status = plyr::mapvalues(rownames(expression_df),
from=bloodBLD2022016033.suppl3$UPN.id,
to=bloodBLD2022016033.suppl3$OS_status))
rownames(annotation) <- rownames(expression_df)
annotation$EFS_status <- factor(plyr::mapvalues(rownames(expression_df),
from=bloodBLD2022016033.suppl3$UPN.id,
to=bloodBLD2022016033.suppl3$OS_status), levels=c(0,1))
annotation$OS_status <- factor(annotation$OS_status, levels=c(0,1))
expression_df_normalized <- scale(expression_df)
expression_df_normalized <- as.data.frame(expression_df_normalized)
expression_df_normalized <- expression_df_normalized[order(expression_df_normalized$ZXSL.MK.other, decreasing = TRUE),]
expression_df_normalized[expression_df_normalized > 2] <- 2
expression_df_normalized[expression_df_normalized < -2] <- -2
options(repr.plot.width=15, repr.plot.height=12)
color_mapping <- colorRamp2(
breaks = c(-2, 0, 2),
colors = c("navy", "white", "firebrick3")
)
# 生成具体的颜色向量(例如 100 个颜色梯度)
my_palette <- color_mapping(seq(-2, 2, length.out = 100))
OS_color <- c("#eeeeee", "#434348")
names(OS_color) <- c('0','1')
EFS_color <- c("#eeeeee", "#434348")
names(EFS_color) <- c('0','1')
annotation_colors <- list(Group = SNF_color,
OS_status = OS_color,
EFS_status = EFS_color)
plot <- pheatmap(t(expression_df_normalized),
annotation_col = annotation,
scale = "none", # 不进行标准化
show_rownames = TRUE,
show_colnames = FALSE,
cluster_rows = FALSE,
cluster_cols = FALSE,
border_color = NA,
color = my_palette,
annotation_colors = annotation_colors,
treeheight_row = 0,
width = 8,
height= 5,
file="Output/Figure5/blood.heatmap.markerGenes.pdf")