2 Figure 1
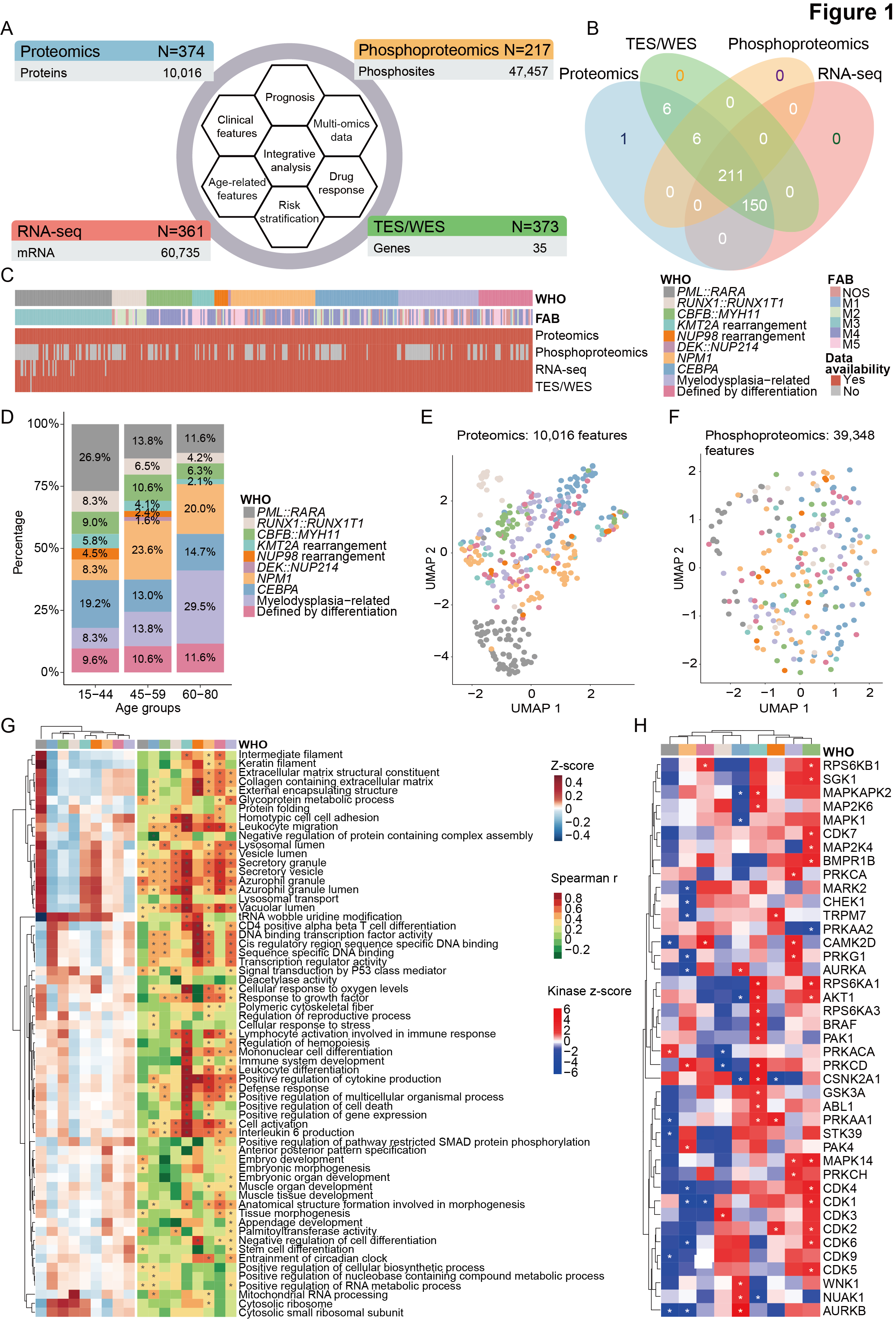
Figure 1. Overview of the study cohort.
A. Schematic overview of multi-omics data obtained from our cohort. Targeted exome sequencing (TES) and whole exome sequencing (WES) were performed on 362 and 11 AML samples, respectively. The analysis focuses on 35 genes that are most frequently mutated in AML.
B. Venn diagram depicting the number of samples within each data layer.
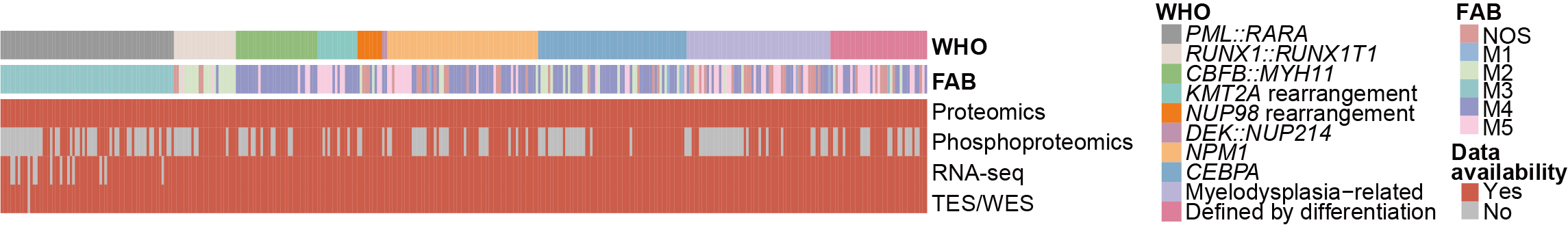
C. The French-American-British (FAB) subtyping, World Health Organization (WHO) classification, and multi-omics information of 374 AML patients.
D. The positive rate of common and other rare gene fusion events in different age groups. Considering the sample size and the balance of data, all patients were categorized into three age groups, namely, 15-44 years, 45-59 years, and 60-80 years.
E-F. Uniform Manifold Approximation and Projection (UMAP) of AML samples using 10,016 measured proteins (E) and 39,348 measured phosphosites (F), with colors denoting different WHO subtypes.
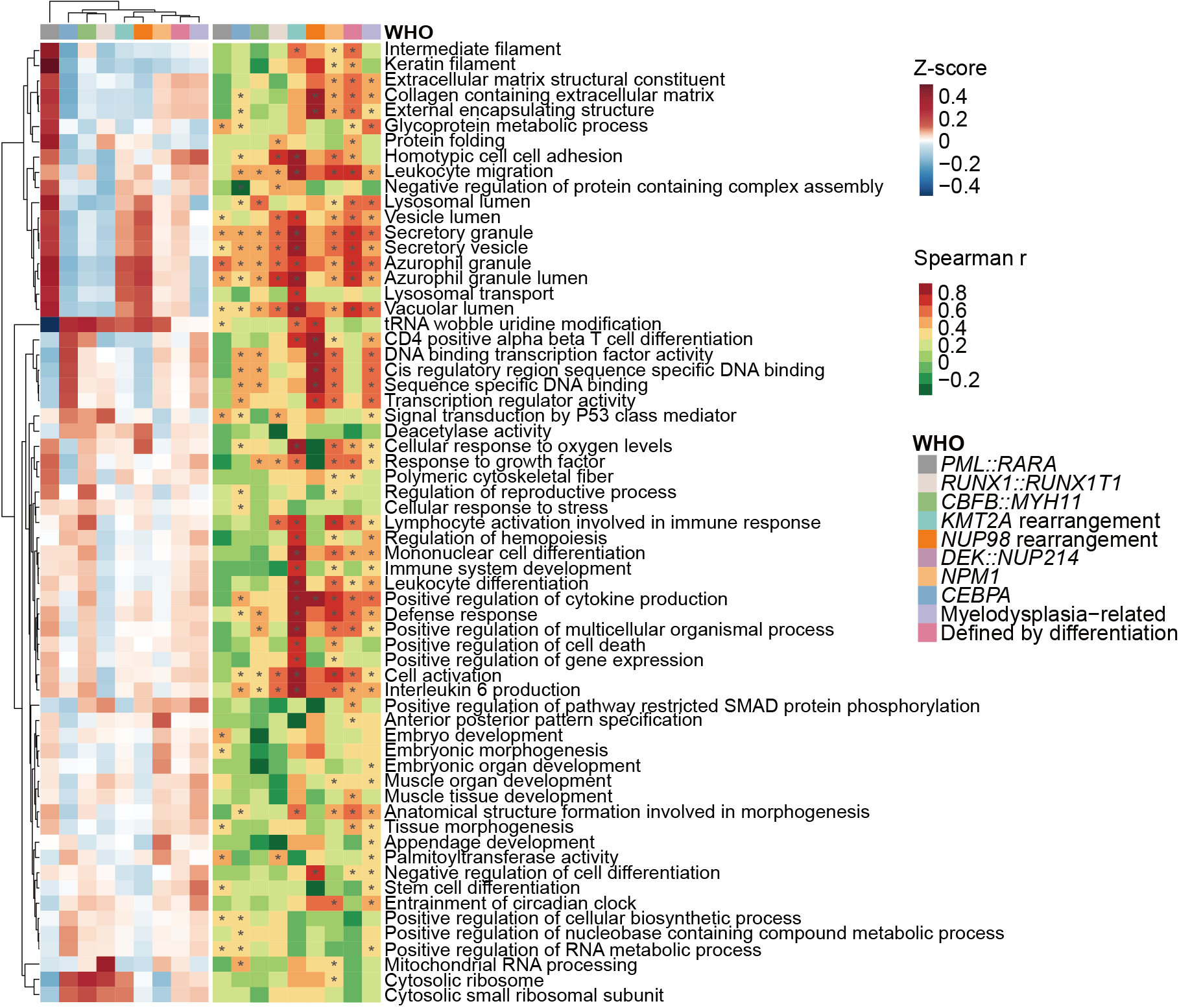
G. Gene set variation analysis (GSVA) scores for proteomics based on differentially regulated gene ontology (GO) pathways in WHO subtypes (left heatmap). Spearman correlation coefficients of the proteome and the transcriptome for the annotated pathways (right heatmap, asterisks mark significantly correlated pathways with P < 0.05, Benjamini-Hochberg-corrected).
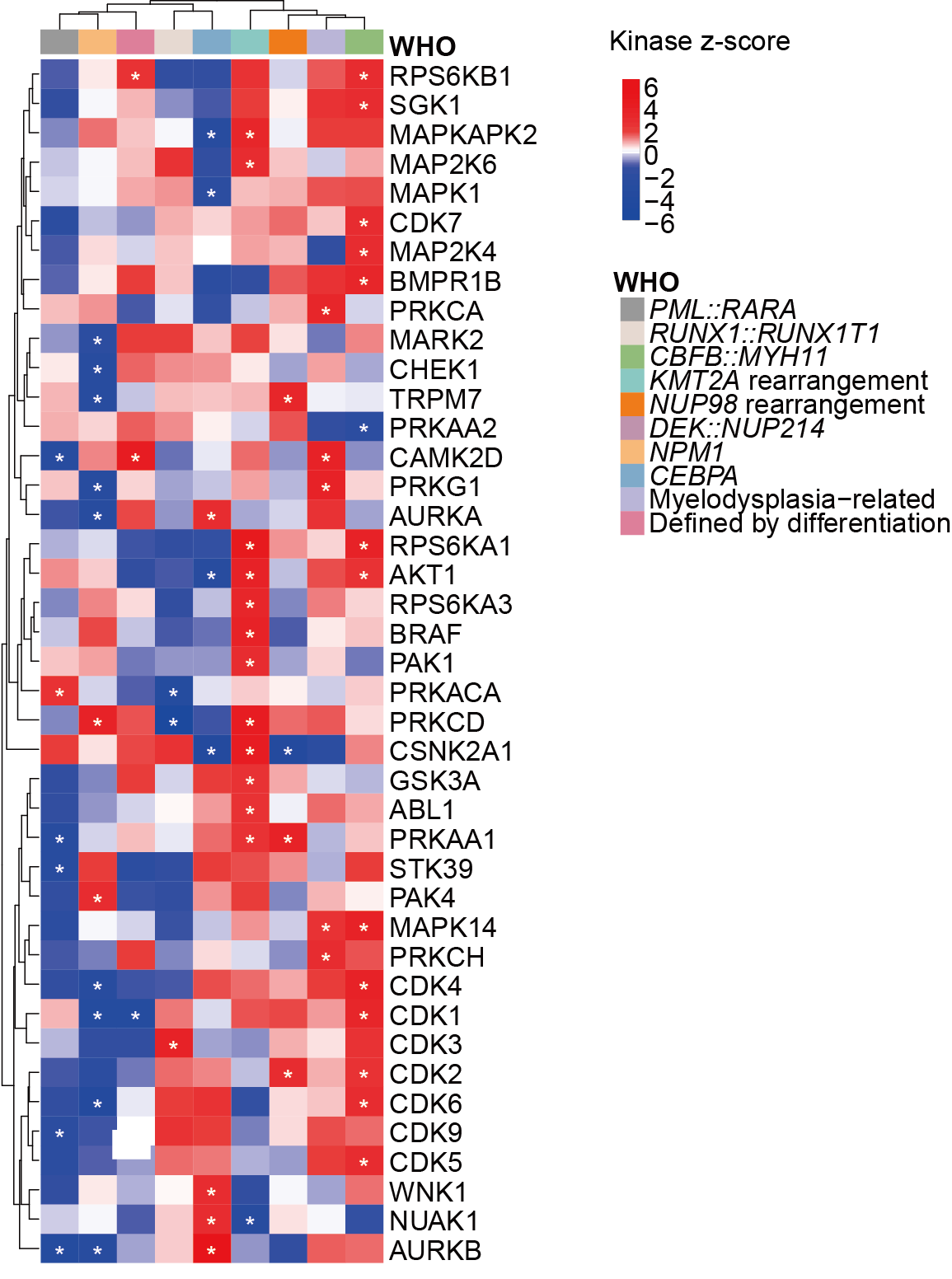
H.Heatmap depicting kinome profiling with Z scores calculated by kinase-substrate enrichment analysis (KSEA) algorithm.
2.1 (B) Data layer
Venn diagram depicting the number of samples within each data layer.
rm(list=ls())
library(VennDiagram)
#-----------------------------------------------------------------------------
#Step 1: Load multi-omics data and sample information
#-----------------------------------------------------------------------------
AML_data <- readRDS("Input/AML_data.rds")
sampleinfo<-AML_data$sampleinfo
all.sample.name<-AML_data$sampleinfo$DIA_ID
prot.sample.name<-names(AML_data$CorrectProteomics)[names(AML_data$CorrectProteomics) %in% all.sample.name]
phospho.sample.name<-names(AML_data$Phosphoproteomics)[names(AML_data$Phosphoproteomics) %in% all.sample.name]
TES_WES.sample.name<-names(AML_data$TES_WES)[names(AML_data$TES_WES) %in% all.sample.name][-317]
RNA_seq.sample.name<-names(AML_data$RNA_seq)[names(AML_data$RNA_seq) %in% all.sample.name]
#-----------------------------------------------------------------------------
#Step 2: Venn Diagram
#-----------------------------------------------------------------------------
venn.plot <- venn.diagram(
x = list("Proteomics"=prot.sample.name,"RNA_seq"=RNA_seq.sample.name,"TES_WES"=TES_WES.sample.name,"Phosphoproteomics"=phospho.sample.name),
filename = NULL,
col = "transparent",
fill = c("#8ebfd5", "#ee8076", "#79c06c", "#f7bc6a"),
alpha = 0.50,
label.col = c("orange", "white", "darkorchid4", "white",
"white", "white", "white", "white", "darkblue", "white",
"white", "white", "white", "darkgreen", "white"),
cex = 1.5,
cat.col = c("black", "black", "black", "black"),
cat.cex = 1.5,
cat.pos = 0,
cat.dist = 0.07,
cat.fontfamily = "serif",
rotation.degree = 0,
margin = 0.2
)
pdf("Output/Figure1/Figure1B.pdf")
grid.draw(venn.plot)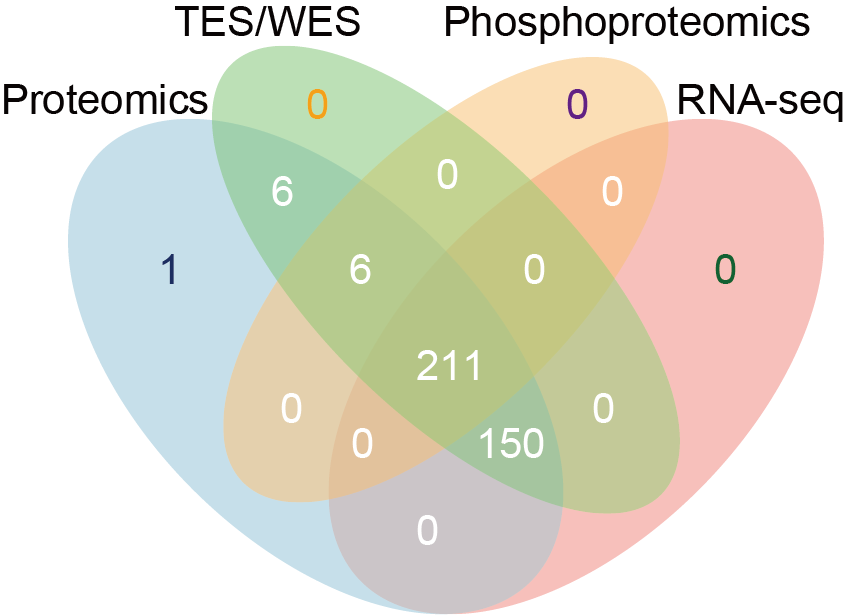
2.2 (C) Data overview
The French-American-British (FAB) subtyping, World Health Organization (WHO) classification, and multi-omics information of 374 AML patients.
rm(list=ls())
library(readxl)
#-----------------------------------------------------------------------------
#Step 1: Load multi-omics data and sample information
#-----------------------------------------------------------------------------
AML_data <- readRDS("Input/AML_data.rds")
sampleinfo<-AML_data$sampleinfo
all.sample.name<-AML_data$sampleinfo$DIA_ID
prot.sample.name<-names(AML_data$CorrectProteomics)[names(AML_data$CorrectProteomics) %in% all.sample.name]
phospho.sample.name<-names(AML_data$Phosphoproteomics)[names(AML_data$Phosphoproteomics) %in% all.sample.name]
TES_WES.sample.name<-names(AML_data$TES_WES)[names(AML_data$TES_WES) %in% all.sample.name][-317]
RNA_seq.sample.name<-names(AML_data$RNA_seq)[names(AML_data$RNA_seq) %in% all.sample.name]
all.ID<- matrix(NA, nrow = length(all.sample.name), ncol = 4, dimnames = list(all.sample.name, c("Proteomics", "Phosphoproteomics", "RNA_seq","TES_WES")))
all.ID[match(prot.sample.name, all.sample.name), "Proteomics"] <- prot.sample.name
all.ID[match(phospho.sample.name, all.sample.name), "Phosphoproteomics"] <- phospho.sample.name
all.ID[match(RNA_seq.sample.name, all.sample.name), "RNA_seq"] <- RNA_seq.sample.name
all.ID[match(TES_WES.sample.name, all.sample.name), "TES_WES"] <- TES_WES.sample.name
all.ID[!is.na(all.ID)] <- 1
all.ID[is.na(all.ID)] <- 0
all.ID<-as.data.frame(all.ID)
all.ID.plot<-data.frame(apply(all.ID, 2, function(x) as.numeric(x)))
rownames(all.ID.plot)<-rownames(all.ID)
all.info<-sampleinfo[,c("DIA_ID", "WHO_classification_2022", "Diagnosis" )]
names(all.info)[2:3]<-c("WHO","FAB")
#-----------------------------------------------------------------------------
#Step 2: Heatmap
#-----------------------------------------------------------------------------
##annotation for heatmap
annotation_col <- data.frame(
FAB=factor(all.info$FAB),
WHO = factor(all.info$WHO),
row.names = all.info$DIA_ID
)
annotation_col <- annotation_col[order(annotation_col$WHO), ]
annotation_col$WHO <- factor(annotation_col$WHO, levels = c(
"PML-RARA", "RUNX1::RUNX1T1", "CBFB::MYH11", "KMT2A rearrangement",
"NUP98 rearrangement", "DEK::NUP214", "NPM1", "CEBPA",
"Myelodysplasia-related", "defined by differentiation"
))
WHO_color<- c("#999999" , "#e7dad2" , "#96c37d", "#8ecfc9","#FF7F00", "#c497b2","#ffbe7a", "#82b0d2" ,"#beb8dc", "#e984a2")
names(WHO_color) <- c(
"PML-RARA","RUNX1::RUNX1T1","CBFB::MYH11","KMT2A rearrangement","NUP98 rearrangement", "DEK::NUP214","NPM1","CEBPA","Myelodysplasia-related", "defined by differentiation"
)
FAB_color <- c("#DF9E9B","#99BADF","#D8E7CA","#99CDCE","#999ACD","#FFD0E9")
names(FAB_color) <- c("AML","M1","M2","M3","M4","M5")
ann_colors <- list(WHO = WHO_color,FAB=FAB_color)
color_series<-c("grey","#D6604D")
all.ID.plot<-all.ID.plot[row.names(annotation_col),]
plot<- pheatmap::pheatmap(
t(all.ID.plot),
scale = "none",
annotation_col = annotation_col,
annotation_colors = ann_colors,
color = color_series,
fontsize_col = 1,
cluster_rows = F,
cluster_cols = F,
show_rownames = T,
show_colnames = F,
fontsize = 10,
cellwidth = 2,
cellheight = 10,
filename =
"Output/Figure1/Figure1C.pdf",
width = 15,
height = 8
)
2.3 (D) Stacked bar chart
The positive rate of common and other rare gene fusion events in different age groups. Considering the sample size and the balance of data, all patients were categorized into three age groups, namely, 15-44 years, 45-59 years, and 60-80 years.
rm(list=ls())
library(plyr)
#-----------------------------------------------------------------------------
#Step 1: Load sample information
#-----------------------------------------------------------------------------
AML_data <- readRDS("Input/AML_data.rds")
data<-as.data.frame(AML_data$sampleinfo)
data$Age_group <- ifelse(data$Age < 45,"15-44","45-59")
data$Age_group[data$Age >= 60] <- "60-80"
data$Age_group <- factor(data$Age_group, levels = c("15-44","45-59","60-80"))
Age_WHO <- data.frame(table(data$Age_group,data$WHO_classification_2022))
Age_WHO <- ddply(Age_WHO,.(Var1),transform,percent=Freq/sum(Freq)*100)
Age_WHO$label = paste0(sprintf("%.1f", Age_WHO$percent), "%")
table(Age_WHO$Var2)
Age_WHO$Var2 <- factor(Age_WHO$Var2, levels = c("PML-RARA","RUNX1::RUNX1T1","CBFB::MYH11",
"KMT2A rearrangement","NUP98 rearrangement",
"DEK::NUP214","NPM1","CEBPA",
"Myelodysplasia-related",
"defined by differentiation"))
#-----------------------------------------------------------------------------
#Step 2: Stacked bar chart
#-----------------------------------------------------------------------------
ggplot(Age_WHO,aes(Var1,percent,fill=Var2))+
geom_bar(stat="identity",position = position_stack())+
labs(x="Age groups",y="Percentage",fill="WHO classification") +
scale_fill_manual(values = c("#999999" , "#e7dad2" , "#96c37d", "#8ecfc9","#FF7F00", "#c497b2","#ffbe7a", "#82b0d2" ,"#beb8dc", "#e984a2"))+
scale_y_continuous(labels = scales::percent_format(scale = 1))+
theme_classic()+
theme(legend.position = "right",
legend.text = element_text(size=10),
axis.text = element_text(size=10),
axis.title = element_text(size=10))+
geom_text(aes(label = sprintf("%1.1f%%", percent)),
stat = "identity",
position = position_stack(vjust = 0.5),
size = 3)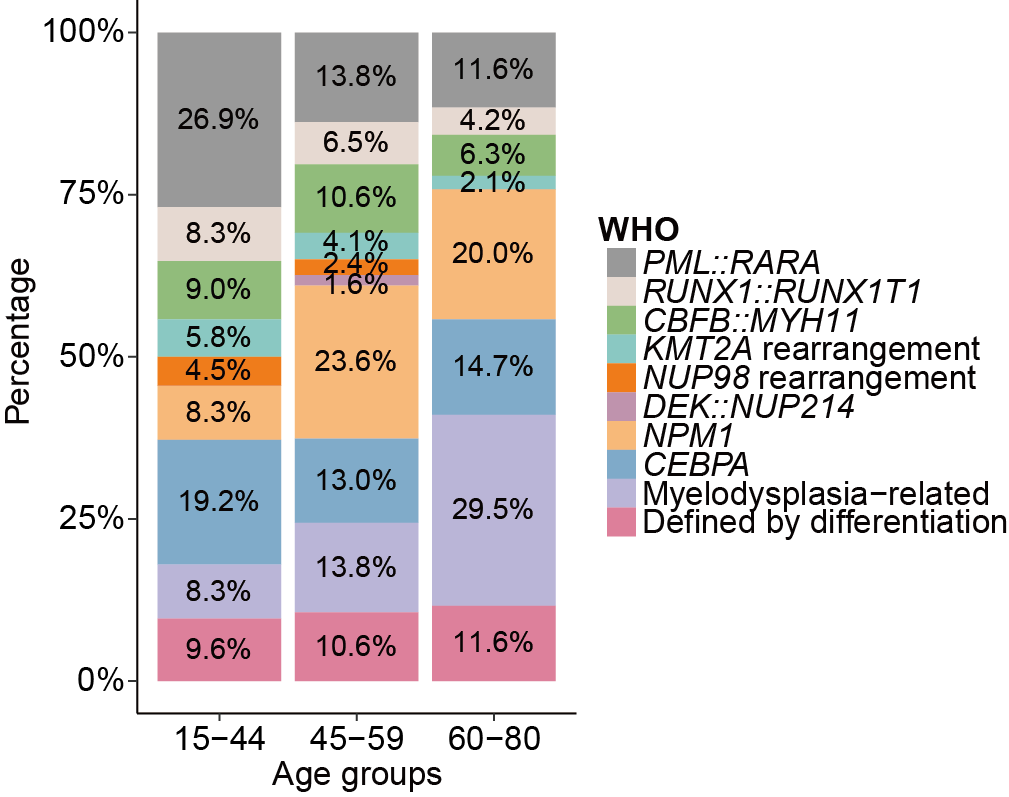
2.4 (E) UMAP for proteomics data
Uniform Manifold Approximation and Projection (UMAP) of AML samples using 10,017 measured proteins.
rm(list = ls())
library(readxl)
library(stringr)
library(umap)
library(RColorBrewer)
library(ggplot2)
#--------------------------------------------------------------------------------
#Step 1: Load proteomics data
#--------------------------------------------------------------------------------
AML_data<-readRDS("Input/AML_data.rds")
prot<-AML_data$CorrectProteomics
info<-AML_data$sampleinfo
prot1<-prot[,-c(1:2)]
prot2<-data.frame(t(prot1))
prot2$DIA_ID<-row.names(prot2)
prot3<-merge(info,prot2,by="DIA_ID",all = F)
prot_plot<-data.frame(prot3[,-c(1:198)])
prot_plot$label<-prot3$WHO_classification_2022
prot_plot<-prot_plot[,-c(1,2,4)]
row.names(prot_plot)<-prot3$DIA_ID
#-------------------------------------------------------------------------------
# Step 2: UMAP
#-------------------------------------------------------------------------------
drawUMAP <- function(M1,ptColors, strTitle="UMAP",rowNormalization=T,colNormalization=F){
if(!'label' %in% colnames(M1)){
print('A column with named label must existed in data frame')
return(NULL)
}
tmp <- M1[,colnames(M1)!='label']
if(rowNormalization){
tmp <- data.frame(t(apply(tmp,1,function(v){(v-mean(v,na.rm=T))/sd(v,na.rm=T)})),stringsAsFactors=F)
rownames(tmp) <- rownames(M1)
}
if(colNormalization){ tmp <- apply(tmp,2,function(v){(v-mean(v,na.rm=T))/sd(v,na.rm=T)}) }
tmp[is.na(tmp)] <- 0
obj = umap(d=tmp,method='naive')
clnames <- colnames(tmp)
df1 <- data.frame(obj$layout)
df1$label <- M1$label
colnames(df1) <- c('X','Y','label')
p <- ggplot(df1, aes(x=X, y=Y, colour=label)) + geom_point(size=4)
p <- p + theme( panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.border = element_blank(),
plot.title = element_text(size=16),
axis.line.x = element_line(color="black", size = 0.5),
axis.line.y = element_line(color="black", size = 0.5),
panel.background = element_blank())
p <- p + labs(title =strTitle)
p <- p + scale_colour_manual(values=ptColors)
p
}
color_order<-c(
"PML-RARA","RUNX1::RUNX1T1","CBFB::MYH11","KMT2A rearrangement","NUP98 rearrangement", "DEK::NUP214","NPM1","CEBPA","Myelodysplasia-related", "defined by differentiation"
)
prot_plot$label<-factor(prot_plot$label,levels = color_order)
color<- c("#999999" , "#e7dad2" , "#96c37d", "#8ecfc9","#FF7F00", "#c497b2","#ffbe7a", "#82b0d2" ,"#beb8dc", "#e984a2")
names(color) <- color_order
set.seed(2024)
prot_umap<-drawUMAP(prot_plot,rowNormalization = F,colNormalization = T,ptColors = color)
ggsave("Output/Figure1/Figure1E.pdf",plot =prot_umap,device = NULL )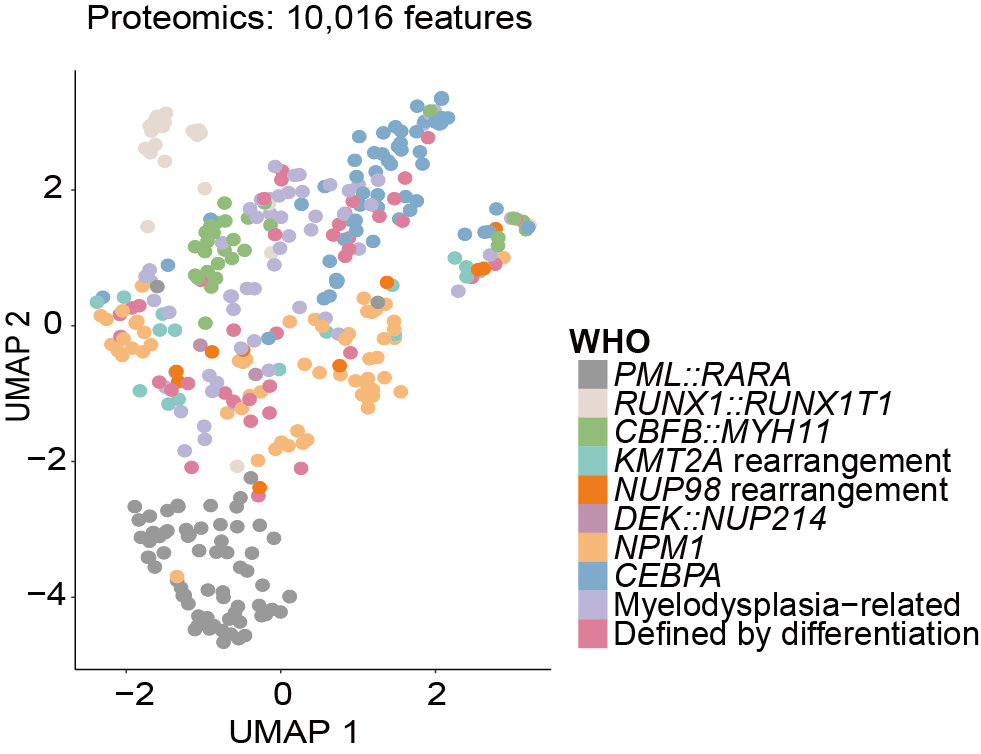
2.5 (F) UMAP for phosphoproteomics data
Uniform Manifold Approximation and Projection (UMAP) of AML samples using 47,457 measured phosphorylated peptides.
#--------------------------------------------------------------------------------
#Step 1: Load phosphoproteomics data
#--------------------------------------------------------------------------------
AML_data<-readRDS("Input/AML_data.rds")
phospho<-AML_data$Phosphoproteomics
info<-AML_data$sampleinfo
row.names(phospho)<-paste(phospho$PTM.FlankingRegion,phospho$PG.ProteinAccessions,phospho$PTM.SiteAA,phospho$PTM.SiteLocation,sep = "-")
phospho1<-phospho[,-c(1:6)]
phospho2<-data.frame(t(phospho1))
phospho2[is.na(phospho2)]<-1
phospho2<-log2(phospho2)
phospho2$DIA_ID<-row.names(phospho2)
phospho3<-merge(info,phospho2,by="DIA_ID",all = F)
phospho_plot<-data.frame(phospho3[,-c(1:198)])
phospho_plot$label<-phospho3$WHO_classification_2022
row.names(phospho_plot)<-phospho3$DIA_ID
WHO<- as.factor(unique(phospho_plot$label))
#-------------------------------------------------------------------------------
# Step 2: UMAP
#-------------------------------------------------------------------------------
phospho_plot$label<-factor(phospho_plot$label,levels = color_order)
phospho_plot<-phospho_plot[,-c(1,2,4)]
set.seed(2023)
phospho_umap<-drawUMAP(phospho_plot,rowNormalization = F,colNormalization = T,ptColors = color)
ggsave("Output/Figure1/Figure1F.pdf",plot =phospho_umap,device = NULL )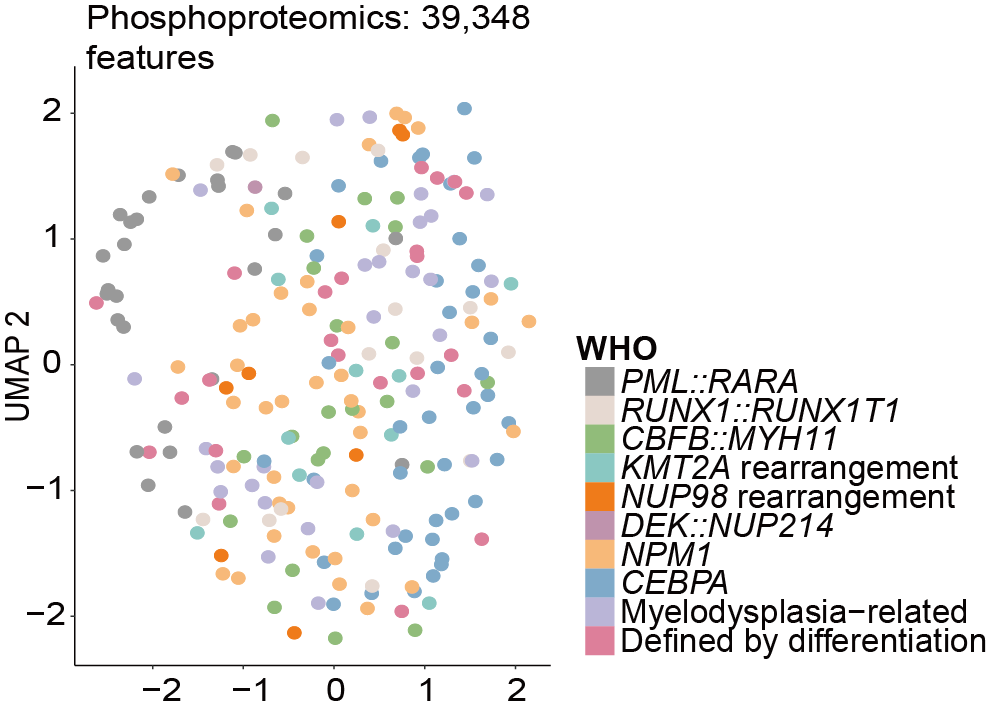
2.6 (G) Gene set variation analysis
Gene set variation analysis (GSVA) scores for proteomics based on differentially regulated gene ontology (GO) pathways in WHO subtypes (left heatmap). Spearman correlation coefficients of the proteome and the transcriptome for the annotated pathways (right heatmap, asterisks mark significantly correlated pathways with P < 0.05, Benjamini-Hochberg-corrected).
rm(list=ls())
library(limma)
library(readxl)
library(dplyr)
library(readr)
library(clusterProfiler)
options(stringsAsFactors = F)
library(tidyverse)
library(msigdbr)
library(GSVA)
library(GSEABase)
library(pheatmap)
library(BiocParallel)
library(RColorBrewer)
#-----------------------------------------------------------------------------
#Step 1: Load data and and set parameters
#-----------------------------------------------------------------------------
AML_data<-readRDS("Input/AML_data.rds")
sample_info<-AML_data$sampleinfo
prot0<-AML_data$CorrectProteomics
prot1<-prot0[-grep(";",prot0$PG.ProteinGroups),]
aml.prot<-data.frame(t(prot1[,-c(1:2)]))
names(aml.prot)<-prot1$PG.Genes
GO_df_C5 <- msigdbr(species = "Homo sapiens",
category = "C5")
GO_df_all<-GO_df_C5
GO_df <- dplyr::select(GO_df_all, gs_name, gene_symbol, gs_exact_source, gs_subcat)
GO_df <- GO_df[GO_df$gs_subcat!="HPO",]
go_list <- split(GO_df$gene_symbol, GO_df$gs_name)
geneset <- go_list
dat<-data.frame(t(aml.prot))
dat<-as.matrix(dat)
#-----------------------------------------------------------------------------
#Step 2: Gene set enrichment analysis
#-----------------------------------------------------------------------------
Pathways<-data.frame("topPathways","group",check.names = F)
names(Pathways)<-c("topPathways","group")
Pathways<-Pathways[-1,]
WHO<-data.frame("DIA_ID"=sample_info$DIA_ID,"WHO"=sample_info$WHO_classification_2022)
WHO$WHO<-gsub(" ","_",WHO$WHO)
WHO$WHO<-gsub("::","_",WHO$WHO)
WHO$WHO<-gsub("-","_",WHO$WHO)
row.names(WHO)<-WHO$DIA_ID
WHO<-WHO[colnames(dat),]
for (i in unique(WHO$WHO)[-10]) {
set.seed(2024)
cluster<-WHO
cluster$WHO<-ifelse(cluster$WHO == i,cluster$WHO,"others")
group_list<-cluster$WHO
design <- model.matrix(~0+factor(group_list))
colnames(design) <- levels(factor(group_list))
rownames(design) <- colnames(dat)
contrast.matrix <- makeContrasts(contrasts=paste0(i,'-',"others"),
levels = design)
fit1 <- lmFit(dat,design)
fit2 <- contrasts.fit(fit1, contrast.matrix)
efit <- eBayes(fit2)
summary(decideTests(efit,lfc=1, p.value=0.05))
tempOutput <- topTable(efit, coef=paste0(i,'-',"others"), n=Inf)
degs0 <- na.omit(tempOutput)
degs0$group<- paste0(i,'-',"others")
degs0<-degs0 %>%
filter(abs(logFC)> 0.5)%>%
filter(adj.P.Val< 0.05)
mydata <-degs0[order(degs0$logFC,decreasing=TRUE),]
FCgenelist <-mydata$logFC
names(FCgenelist)<-row.names(mydata)
fgseaRes <- fgsea::fgsea(pathways= geneset,
stats= FCgenelist,
minSize=10,
maxSize=500,
nperm=10000, nproc = 1)
fgseaRes<-fgseaRes %>%
filter(pval<0.05)
topPathwaysUp.tmp <- fgseaRes[NES > 0]
topPathwaysDown.tmp <- fgseaRes[NES < 0]
if (length(topPathwaysUp.tmp) < 5 ){
up= length(topPathwaysUp.tmp)
} else{
up=5
}
if (length(topPathwaysDown.tmp) <5 ){
down= length(topPathwaysDown.tmp)
} else{
down=5
}
topPathwaysUp <- fgseaRes[NES > 0][head(order(NES,decreasing = T),n=5),pathway]
topPathwaysDown <- fgseaRes[NES < 0][head(order(NES,decreasing = F),n=5),pathway]
topPathways <-c(topPathwaysUp,rev(topPathwaysDown))
if (length(topPathways)== 0){
topPathways<-"NA"
}else{
topPathways<-topPathways
}
Pathways0<-data.frame(topPathways,"group")
Pathways0$X.group.<-paste0(i,'-',"others")
names(Pathways0)[2]<-"group"
Pathways<-rbind(Pathways,Pathways0)
}
geneset.filter<-geneset[names(geneset) %in% Pathways$topPathways]
#-----------------------------------------------------------------------------
#Step 3: Gene set variation analysis
#-----------------------------------------------------------------------------
prot.data<-dat
set.seed(123)
param <- gsvaParam(exprData = prot.data,
geneSets = geneset.filter,
kcdf = "Gaussian")
gsva.prot <- gsva(param,
verbose = TRUE,
#parallel.sz = parallel::detectCores()
)
degs<-data.frame("logFC", "AveExpr","t" ,"P.Value","adj.P.Val" ,"B", "group", "pathway")
names(degs)<-c("logFC", "AveExpr","t" ,"P.Value","adj.P.Val" ,"B", "group", "pathway")
degs<-degs[-1,]
for (i in unique(WHO$WHO)[-10]) {
set.seed(123)
cluster<-WHO
cluster$WHO<-ifelse(cluster$WHO == i,cluster$WHO,"others")
group_list<-cluster$WHO
design <- model.matrix(~0+factor(group_list))
colnames(design) <- levels(factor(group_list))
rownames(design) <- colnames(gsva.prot)
contrast.matrix <- makeContrasts(contrasts=paste0(i,'-',"others"),
levels = design)
fit1 <- lmFit(gsva.prot,design)
fit2 <- contrasts.fit(fit1, contrast.matrix)
efit <- eBayes(fit2)
tempOutput <- topTable(efit, coef=paste0(i,'-',"others"), n=Inf)
degs0 <- na.omit(tempOutput)
degs0$group<- paste0(i,'-',"others")
degs0$pathway<-row.names(degs0)
degs<-rbind(degs0,degs)
}
degs.del.snf5<-degs
library(reshape2)
pathway.heatmap<- dcast(degs.del.snf5[,c(1,7,8)], pathway ~ group, value.var = "logFC")
pathway.heatmap.1<-pathway.heatmap[,-1]
row.names(pathway.heatmap.1)<-pathway.heatmap$pathway
colors<- c('#053061','#0F437C','#195797','#246AAE','#307AB6','#3C8ABE','#519CC7','#6EAED1','#8AC0DB','#A3CDE2','#BAD9E9','#D1E5F0',"white", "#FDDBC7", "#F4A582","#D6604D", "#C94E45", "#C23D3E", "#BA2C37", "#B21C30", "#A90B29", "#A10328", "#920226", "#820224", "#67001F")
color_series <- colorRampPalette(colors)(1000)
heatmap <- pheatmap::pheatmap(
pathway.heatmap.1,
color = color_series,
fontsize_col = 4,
cluster_rows = T,
cluster_cols = T,
show_rownames = T,
show_colnames = T,
fontsize = 2,
cellwidth = 10,
cellheight = 2,
filename =paste0("Output/Figure1/Figure1G_1.pdf"))
all.prot<-names(aml.prot)
prot.in.genenset<-data.frame(1:5000)
for (i in row.names(pathway.heatmap.1)) {
prot.in.genenset0<- geneset.filter[[i]][geneset.filter[[i]] %in% all.prot]
prot.in.genenset0<-data.frame(prot.in.genenset0)
names(prot.in.genenset0)<-i
row<- nrow(prot.in.genenset0)+1
prot.in.genenset0[row:5000,]<-NA
prot.in.genenset<- cbind (prot.in.genenset, prot.in.genenset0)
}
prot.in.genenset <- prot.in.genenset[, -1]
row_na_percentage <-
rowSums(is.na(prot.in.genenset)) / ncol(prot.in.genenset)
threshold <- 0.999999
selected_rows <- which(row_na_percentage < threshold)
prot.in.genenset.delte.na <- prot.in.genenset[selected_rows,]
trans<-AML_data$RNA_seq
trans.data<-as.matrix((trans[,-c(1:2)]))
row.names(trans.data)<-trans$symbol
trans.data<-data.frame(trans.data)
trans.data<-trans.data[,names(trans.data) %in% row.names(WHO)]
trans.data<-as.matrix(trans.data)
set.seed(123)
param2<-gsvaParam(exprData = trans.data,
geneSets = geneset.filter,
kcdf = "Gaussian")
gsva.trans <- gsva(param2,
verbose = TRUE)
#parallel.sz = parallel::detectCores())
gsva.trans<-data.frame(gsva.trans)
gsva.prot<-data.frame(gsva.prot)
gsva.prot<-gsva.prot[,names(gsva.trans)]
trans.data<-data.frame(gsva.trans)
prot.data<-data.frame(gsva.prot)
df<-data.frame("corr","corr_p","pathway","cluster")
names(df)<-c("corr","corr_p","pathway","cluster")
df<-df[-1,]
for (cluster in unique(WHO$WHO)[-10]) {
cluster.sample <- row.names(WHO[WHO$WHO == cluster, ])
trans.cluster <-
trans.data[, names(trans.data) %in% cluster.sample]
prot.cluster <- prot.data[, names(prot.data) %in% cluster.sample]
for (i in 1:nrow(trans.cluster)) {
pathway <- row.names(trans.cluster)[i]
trans.pathway <- c(as.matrix(trans.cluster[i, ]))
prot.pathway <- c(as.matrix(prot.cluster[i, ]))
corr <- cor(trans.pathway, prot.pathway, method = "spearman")
corr_p <-cor.test(trans.pathway, prot.pathway, method = "spearman")$p.value
df0 <- data.frame(corr, corr_p, pathway, cluster, check.names = F)
df <- rbind(df0, df)
}
}
row.order<- heatmap$tree_row$order
col.order<-heatmap$tree_col$order
corr.heatmap<- dcast(df, pathway ~ cluster, value.var = "corr")
corr.heatmap.1<-corr.heatmap[,-c(1)]
row.names(corr.heatmap.1)<-corr.heatmap$pathway
corr.heatmap.1<-corr.heatmap.1[row.order,col.order]
corr.heatmap.p<- dcast(df, pathway ~ cluster, value.var = "corr_p")
corr.heatmap.p.1<- corr.heatmap.p[,-c(1)]
row.names(corr.heatmap.p.1)<-corr.heatmap.p$pathway
corr.heatmap.p.1<-corr.heatmap.p.1[row.order,col.order]
significance_level = 0.05
significant_matrix <-ifelse(corr.heatmap.p.1 < significance_level, "*", "")
significant_matrix[is.na(significant_matrix)] <- ""
colors <- brewer.pal(name = "RdYlGn", n = 10)[10:1]
heatmap <- pheatmap::pheatmap(
corr.heatmap.1,
color = colors,
fontsize_col = 4,
display_numbers = significant_matrix,
cluster_rows = F,
cluster_cols = F,
show_rownames = T,
show_colnames = T,
fontsize = 2,
cellwidth = 10,
cellheight = 2,
filename = paste0( "Output/Figure1/Figure1G_2.pdf")
)
2.7 (H) kinase-substrate enrichment analysis
Heatmap depicting kinome profiling with Z scores calculated by kinase-substrate enrichment analysis (KSEA) algorithm.
rm(list=ls())
library(dplyr)
library(MNet)
library(stringr)
library(ggplot2)
library(RColorBrewer)
library(readr)
library(dplyr)
library(MNet)
library(stringr)
library(ggplot2)
library(RColorBrewer)
library(KSEAapp)
#-----------------------------------------------------------------------------
#Step 1: Load data and and set parameters
#-----------------------------------------------------------------------------
AML<-read_rds("Input/AML_data.rds")
info<-AML$sampleinfo
phospho<-AML$Phosphoproteomics
row.names(phospho)<- paste(sep = "_", phospho$PTM.FlankingRegion,phospho$PTM.SiteAA,phospho$PTM.SiteLocation,phospho$PG.Genes)
phospho1<-phospho[,-c(1:6)]
phosphoNA20<-apply(phospho1,1, function (x) {sum(is.na(x))/(ncol(phospho1))})
phospho2<-phospho1[names(phosphoNA20)[phosphoNA20<0.8],]
phospho_name<-names(phospho2)
names(phospho2)<-phospho_name
phospho3<-phospho2
phospho4<-log2(phospho3)
phospho4[is.na(phospho4)]<-0
phospho5<-data.frame(t(phospho4),check.names = F)
phospho5$DIA_ID<-row.names(phospho5)
df2<-merge(info[,c(5,31)],phospho5,by="DIA_ID",all.x=F,all.y = F)
names(df2)[2]<-"WHO"
df3<-data.frame(t(df2[,-c(1:2)]))
names(df3)<-df2$DIA_ID
df2$WHO<-gsub(" ","_",df2$WHO)
df2$WHO<-gsub("::","_",df2$WHO)
df2$WHO<-gsub("-","_",df2$WHO)
matrix_raw<- data.frame(apply(df3,2,function (x) {as.numeric(x)}))
row.names(matrix_raw)<- row.names(df3)
iN<- unique(df2$WHO)[-10]
for (i in iN) {
if ( i %in% iN) {
type <- c(as.matrix(df2$WHO))
type[!type == i] <- "normal"
type[type == i] <- "tumor"
aml_matrix <- matrix_raw
}
#-----------------------------------------------------------------------------
#Step 2: Kinase–substrate enrichment analysis
#-----------------------------------------------------------------------------
diff_phospho_KSEA<- MNet::mlimma(aml_matrix,type)
phosphoname<-data.frame(row.names(phospho),phospho[,c(2,3,1,5,6)])
phosphoname$site<-paste0(phosphoname$PTM.SiteAA,phosphoname$PTM.SiteLocation)
names(phosphoname)[1]<-c("name")
diff_phospho_KSEA1<-merge(diff_phospho_KSEA,phosphoname,by="name",all.x = T,all.y = F)
diff_phospho_KSEA2<-diff_phospho_KSEA1[,c(9:11,14,5,2)]
names(diff_phospho_KSEA2)<-c("Protein","Gene","Peptide","Residue.Both", "p","FC")
diff_phospho_KSEA2$FC<-2^diff_phospho_KSEA2$FC
KSData=read.csv("Input/PSP&NetworKIN_Kinase_Substrate_Dataset_July2016.csv", header = T)
PX=diff_phospho_KSEA2
path<-paste0("Output/Figure1")
KSEA.Complete(KSData,
PX,
NetworKIN = T,
NetworKIN.cutoff = 5,
m.cutoff = 5,
p.cutoff = 0.05)
KSEA.Scores=KSEA.Scores(KSData,
PX,
NetworKIN = T,
NetworKIN.cutoff = 20
)
write.csv(KSEA.Scores, paste0(path,i,"_KSEA.Scores.csv"),row.names = FALSE)
}
CBFB_MYH11<-read.csv(paste0(path,"CBFB_MYH11_KSEA.Scores.csv"))
KMT2A_rearrangement<-read.csv(paste0(path,"KMT2A_rearrangement_KSEA.Scores.csv"))
NPM1<-read.csv(paste0(path,"NPM1_KSEA.Scores.csv"))
defined_by_differentiation<-read.csv(paste0(path,"defined_by_differentiation_KSEA.Scores.csv"))
CEBPA<-read.csv(paste0(path,"CEBPA_KSEA.Scores.csv"))
Myelodysplasia_related<-read.csv(paste0(path,"Myelodysplasia_related_KSEA.Scores.csv"))
RUNX1_RUNX1T1<-read.csv(paste0(path,"RUNX1_RUNX1T1_KSEA.Scores.csv"))
PML_RARA<-read.csv(paste0(path,"PML_RARA_KSEA.Scores.csv"))
NUP98_rearrangement<-read.csv(paste0(path,"NUP98_rearrangement_KSEA.Scores.csv"))
KSEA.Heatmap.new <- function(score.list, sample.labels, stats, m.cutoff, p.cutoff, sample.cluster) {
library(gplots)
filter.m <- function(dataset, m.cutoff) {
filtered <- dataset[(dataset$m >= m.cutoff), ]
return(filtered)
}
score.list.m <- lapply(score.list, function(...) filter.m(..., m.cutoff))
for (i in 1:length(score.list.m)) {
names <- colnames(score.list.m[[i]])[c(2:7)]
colnames(score.list.m[[i]])[c(2:7)] <- paste(names, i, sep = ".")
}
master <- Reduce(function(...) merge(..., by = "Kinase.Gene", all = F), score.list.m)
row.names(master) <- master$Kinase.Gene
columns <- as.character(colnames(master))
merged.scores <- as.matrix(master[, grep("z.score", columns)])
colnames(merged.scores) <- sample.labels
merged.stats <- as.matrix(master[, grep(stats, columns)])
asterisk <- function(matrix) {
new <- data.frame()
for (i in 1:nrow(matrix)) {
for (j in 1:ncol(matrix)) {
if (matrix[i, j] < p.cutoff) {
new[i, j] <- "*"
} else {
new[i, j] <- ""
}
}
}
return(new)
}
merged.asterisk <- as.matrix(asterisk(merged.stats))
row.names(merged.asterisk) <- row.names(master)
p_detect <- data.frame(asterisk(merged.stats), row.names = row.names(master))
p_detect[p_detect != "*"] <- NA
p_detect.1 <- p_detect[!apply(p_detect, 1, function(row) all(is.na(row))), ]
merged.asterisk <- data.frame(merged.asterisk, row.names = row.names(master))
merged.asterisk <- merged.asterisk[row.names(merged.asterisk) %in% row.names(p_detect.1), ]
merged.asterisk <- as.matrix(merged.asterisk)
create.breaks <- function(merged.scores) {
if (min(merged.scores) < -1.6) {
breaks.neg <- seq(-1.6, 0, length.out = 30)
breaks.neg <- append(seq(min(merged.scores), -1.6, length.out = 10), breaks.neg)
breaks.neg <- sort(unique(breaks.neg))
} else {
breaks.neg <- seq(-1.6, 0, length.out = 30)
}
if (max(merged.scores) > 1.6) {
breaks.pos <- seq(0, 1.6, length.out = 30)
breaks.pos <- append(breaks.pos, seq(1.6, max(merged.scores), length.out = 10))
breaks.pos <- sort(unique(breaks.pos))
} else {
breaks.pos <- seq(0, 1.6, length.out = 30)
}
breaks.all <- unique(append(breaks.neg, breaks.pos))
mycol.neg <- colorpanel(n = length(breaks.neg), low = "blue", high = "white")
mycol.pos <- colorpanel(n = length(breaks.pos) - 1, low = "white", high = "red")
mycol <- unique(append(mycol.neg, mycol.pos))
color.breaks <- list(breaks.all, mycol)
return(color.breaks)
}
color.breaks <- create.breaks(merged.scores)
plot.height <- nrow(merged.scores)^0.55
plot.width <- ncol(merged.scores)^0.7
merged.scores <- merged.scores[row.names(merged.scores) %in% row.names(p_detect.1), ]
write.csv(merged.scores, paste0(path, "SNF_KSEA.Merged.Scores.csv"))
pdf(paste0(path, "Figure1H.pdf"), width = 10, height = 30)
heatmap.2(
merged.scores,
Colv = sample.cluster,
scale = "none",
cellnote = merged.asterisk,
notecol = "white",
cexCol = 0.9,
cexRow = 0.9,
srtCol = 45,
notecex = 1.4,
col = color.breaks[[2]],
density.info = "none",
trace = "none",
key = TRUE,
keysize = 0.2,
key.par = list(cex = 0.7),
breaks = color.breaks[[1]],
lmat = rbind(c(4, 3), c(2, 1)),
lhei = c(0.5, 9),
lwid = c(1.5, 4),
margins = c(2, 6)
)
dev.off()
}
KSEA.Heatmap.new(
list(
CBFB_MYH11,
KMT2A_rearrangement,
NPM1,
defined_by_differentiation,
CEBPA,
Myelodysplasia_related,
RUNX1_RUNX1T1,
PML_RARA,
NUP98_rearrangement
),
sample.labels = c(
"CBFB_MYH11",
"KMT2A_rearrangement",
"NPM1",
"defined_by_differentiation",
"CEBPA",
"Myelodysplasia_related",
"RUNX1_RUNX1T1",
"PML_RARA",
"NUP98_rearrangement"
),
stats = 'p.value',
m.cutoff = 3,
p.cutoff = 0.01,
sample.cluster = TRUE
)