4 Figure 3
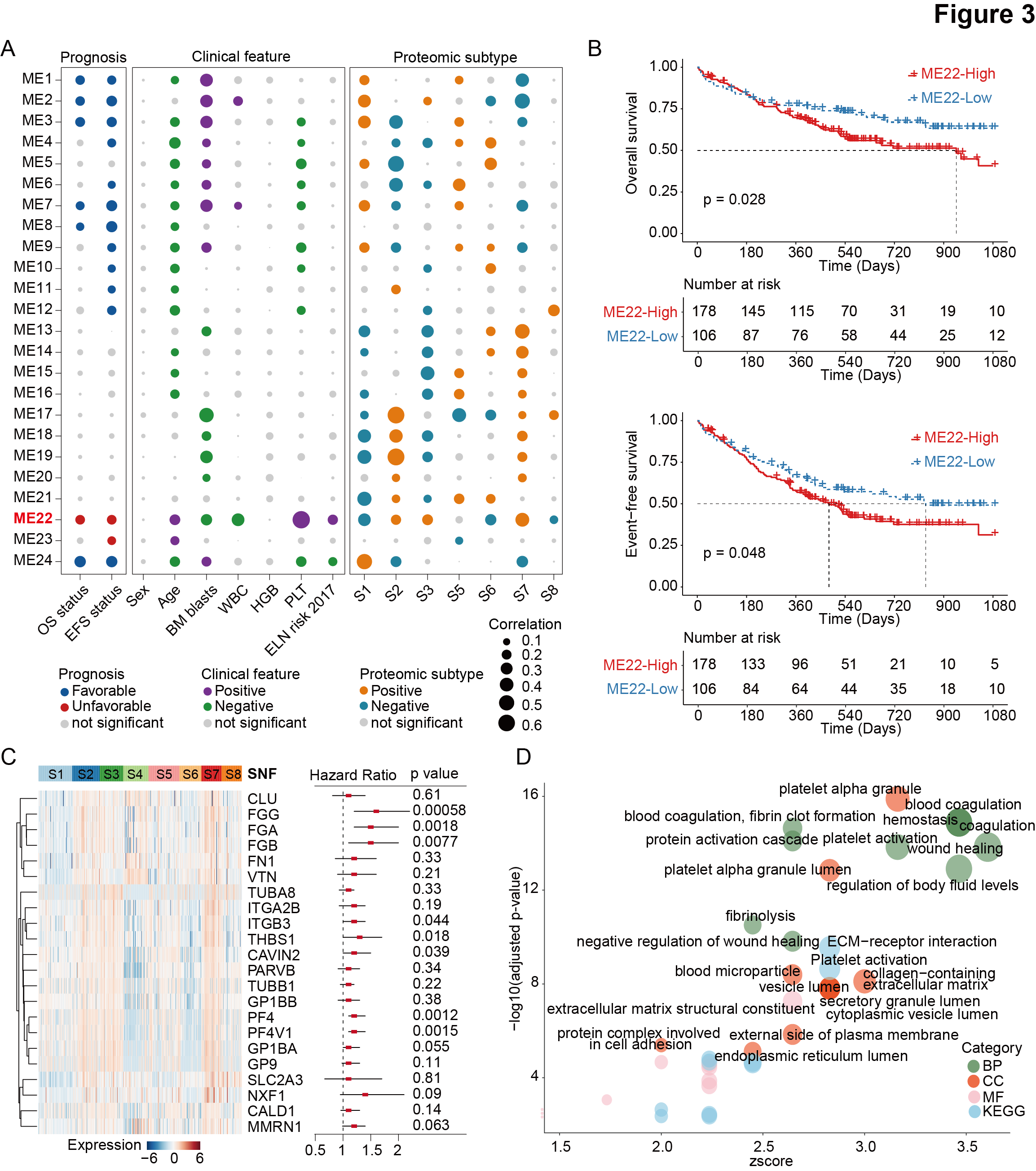
Figure 3.Weighted correlation network analysis identifies functional protein modules with clinical relevance.
Bubble plot displaying 24 protein clusters (functional modules, ME1-24). Modules highly correlated with clinical features, molecular characteristics, and protein expression subgroups are colored. The ME22 is highlighted in red font.
Kaplan-Meier curves for overall survival (upper panel) and event-free survival (lower panel) of AML patients stratified by protein abundance in ME22, with the most obvious prognostic discrimination used as the cutoff value.
Heatmap showing the relative abundance of proteins derived from the ME22 module (left panel). Univariate Cox regression analysis indicating the prognostic value of each protein (right panel). The middle red points indicate the hazard ratio for each protein, and endpoints represent lower or upper 95% confidence intervals (CIs).
Pathway enrichment analysis of ME22-derived proteins between the combination of S2, S3, and S7 and residual AML patients using biological process (BP), cellular component (CC), and molecular function (MF) in Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG).
4.1 (A) Weighted correlation network analysis
Bubble plot displaying 24 protein clusters (functional modules, ME1-24). Modules highly correlated with clinical features, molecular characteristics, and protein expression subgroups are colored. The ME22 is highlighted in red font.
rm(list=ls())
options(stringsAsFactors = FALSE)
library(WGCNA)
library(readxl)
library(dplyr)
library(tidyr)
#-----------------------------------------------------------------------------
#Step 1: Load data and and set parameters
#-----------------------------------------------------------------------------
AML<-read_rds("Input/AML_data.rds")
SNF<-read_rds("Input/SNF_group.rds")
PML<-SNF[SNF$WHO=="PML-RARA",]
SNF<-SNF[SNF$WHO!= PML$WHO,]
SNF<-SNF[SNF$SNF!= "SNF4",]
femData0 <-AML$CorrectProteomics
femData0<-femData0[,!names(femData0) == "T386"]
row.names(femData0)<-femData0$PG.ProteinGroups
femData.raw<-2^femData0[,names(femData0) %in% SNF$DIA_ID]
vec<-apply(femData.raw,1,mean)
Q1 <- quantile(vec, 0.25)
Q3 <- quantile(vec, 0.75)
IQR_value <- Q3 - Q1
lower_threshold <- Q1 - 2 * IQR_value
upper_threshold <- Q3 + 2 * IQR_value
non.outliers <- vec[vec >= lower_threshold & vec <= upper_threshold ]
non.outliers.name<-names(non.outliers)
femData.raw<-femData.raw[row.names(femData.raw) %in% non.outliers.name,]
sd <- apply(femData.raw, 1, function(x) {sd(x)})
sd.top1000<-order(sd, decreasing = TRUE)[1:4000]
femData<-femData.raw[sd.top1000,]
femData<-log2(femData)
datExpr0 = as.data.frame(t(femData))
gsg = goodSamplesGenes(datExpr0, verbose = 3)
gsg$allOK
if (!gsg$allOK)
{
if (sum(!gsg$goodGenes)>0)
printFlush(paste("Removing genes:", paste(names(datExpr0)[!gsg$goodGenes], collapse = ", ")));
if (sum(!gsg$goodSamples)>0)
printFlush(paste("Removing samples:", paste(rownames(datExpr0)[!gsg$goodSamples], collapse = ", ")));
datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}
#-----------------------------------------------------------------------------
#Step 2: Identification of outlier samples
#-----------------------------------------------------------------------------
sampleTree = hclust(dist(datExpr0), method = "average")
cut=200
clust = cutreeStatic(sampleTree, cutHeight = cut, minSize = 10)
table(clust)
keepSamples = (clust==1)
datExpr = datExpr0[keepSamples, ]
#----------------------------------------------------------------------------------
# Step 3: Analysis of network topology
#----------------------------------------------------------------------------------
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
traitData = AML$sampleinfo
library(reshape2)
SNF$value<-1
SNF.wide<-dcast(SNF,DIA_ID ~ SNF, value.var = "value")
traitData<-merge(SNF.wide,traitData,by="DIA_ID",all=F)
dim(traitData)
names(traitData)
allTraits = traitData[, c( "Sex", "Age" ,"BM_blasts" ,"WBC" , "HGB" , "PLT", "ELN_risk_2017" , "biCEBPA", "EFS_status" , "OS_status" , "SNF1" , "SNF2", "SNF3", "SNF5" , "SNF6" , "SNF7","SNF8" )]
allTraits$Sex<-gsub("M","1",allTraits$Sex)
allTraits$Sex<-gsub("F","0",allTraits$Sex)
allTraits<-data.frame(apply(allTraits,2,function (x){as.numeric(x)}))
allTraits[is.na(allTraits)]<-0
row.names(allTraits)<-traitData$DIA_ID
dim(allTraits)
names(allTraits)
Samples = rownames(datExpr)
traitRows = match(Samples,row.names(allTraits))
datTraits = allTraits[traitRows, ]
collectGarbage()
sampleTree2 = hclust(dist(datExpr), method = "average")
traitColors = numbers2colors(datTraits, signed = FALSE)
#----------------------------------------------------------------------------------
# Step 4: Soft threshold : power
#----------------------------------------------------------------------------------
powers = c(c(1:20))
sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
sizeGrWindow(9, 5)
par(mfrow = c(1,2))
cex1 = 0.9
sft$powerEstimate
if (is.na(power)){
power = ifelse(nSamples<20, ifelse(type == "unsigned", 9, 18),
ifelse(nSamples<30, ifelse(type == "unsigned", 8, 16),
ifelse(nSamples<40, ifelse(type == "unsigned", 7, 14),
ifelse(type == "unsigned", 6, 12))
)
)
}
cor<-WGCNA::cor
#----------------------------------------------------------------------------------
# Step 5: One-step network construction and module detection
#----------------------------------------------------------------------------------
net =WGCNA:: blockwiseModules(datExpr, power =6,
TOMType = "unsigned", minModuleSize =20,
reassignThreshold = 0, mergeCutHeight =0.01,
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs = TRUE,
saveTOMFileBase = "AML_Proteomics",
verbose = 3)
moduleLabels = net$colors
moduleColors = labels2colors(net$colors)
MEs = net$MEs
geneTree = net$dendrograms[[1]]
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
MEs0 = moduleEigengenes(datExpr, moduleColors)$eigengenes
MEs = orderMEs(MEs0)
#----------------------------------------------------------------------------------
# Step 6: Vasualization
#----------------------------------------------------------------------------------
moduleTraitCor = cor(MEs, datTraits, use = "p")
moduleTraitPvalue = corPvalueStudent(moduleTraitCor, nSamples);
sizeGrWindow(10,6)
textMatrix = paste(signif(moduleTraitCor, 2), "\n(",
signif(moduleTraitPvalue, 1), ")", sep = "")
dim(textMatrix) = dim(moduleTraitCor)
Cor<-data.frame(moduleTraitCor)
Cor$Module<-paste0("ME",1:nrow(moduleTraitCor))
Cor_long<-gather(Cor,Factor,corr,-Module)
Pvalue<-data.frame(moduleTraitPvalue)
Pvalue$Module<-paste0("ME",1:nrow(moduleTraitCor))
Pvalue_long<-gather(Pvalue,Factor,p,-Module)
Module_trait<-Cor_long
Module_trait$p<-Pvalue_long$p
Module_trait<-Module_trait[!grepl("biCEBPA",Module_trait$Factor),]
Module_trait$Type<- ifelse(grepl("status",Module_trait$Factor), "Prognosis",
ifelse(grepl("SNF",Module_trait$Factor), "SNF", "Clinical"))
Module.bubble<-Module_trait %>%
mutate(Tag=case_when(Type== "Prognosis"&p<0.05& corr>0 ~ "Unfavorable",
Type=="Prognosis" &p<0.05 & corr<0 ~ "Favorable",
Type== "Clinical"&p<0.05& corr<0 ~ "Negative",
Type=="Clinical" &p<0.05 & corr>0 ~ "Positive",
Type== "SNF"&p<0.05& corr<0 ~ "Neg",
Type=="SNF" &p<0.05 & corr>0 ~ "Pos",
.default="notSig" ))
plot.info <- Module.bubble %>% mutate(LogP=-log10(p),
Module=factor(as.character(Module), levels = rev(c("ME1","ME2","ME3","ME4","ME5","ME6","ME7","ME8","ME9","ME10","ME11","ME12","ME13","ME14","ME15","ME16","ME17","ME18","ME19","ME20","ME21","ME22","ME23","ME24","ME25","ME26","ME27"))),
Factor=factor(as.character(Factor), levels = c( "OS_status","EFS_status","Sex","Age","BM_blasts","WBC","HGB","PLT","ELN_risk_2017", "SNF1", "SNF2", "SNF3", "SNF5", "SNF6", "SNF7", "SNF8")))
library(ggplot2)
library(ggthemes)
p <- ggplot(plot.info, aes(Factor, Module)) +
geom_point(aes(size = abs(corr), colour = Tag, fill = Tag), shape = 16) +
xlab("") + ylab("")
p <- p + theme_base() + scale_size(range = c(0,10))
p <- p + theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5, size = 15))
p <- p + theme(axis.text.y = element_text(size = 20))
p <- p + scale_colour_manual(values = c(Favorable = "#00599F", Unfavorable = "#D01910", Negative = "#009632", Positive = "#8f00b7", Pos = "#ed7a00", Neg = "#1288a5", notSig = "#CCCCCC")) + theme(plot.background = element_blank())
ggsave(paste0("Output/Figure3/Figure3A.pdf"), p, width = 12, height = 11)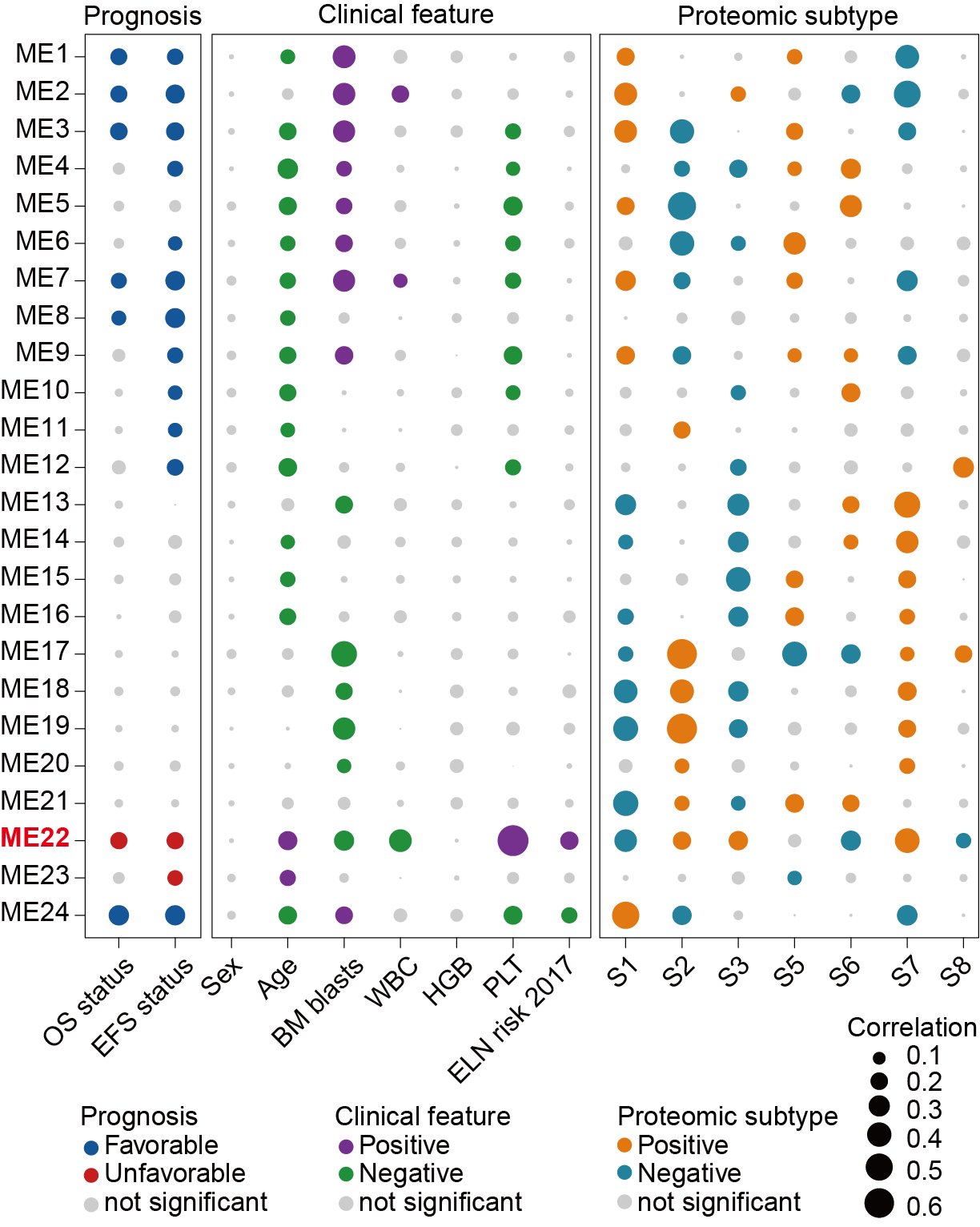
4.2 (B) Kaplan-Meier curves
Kaplan-Meier curves for overall survival (upper panel) and event-free survival (lower panel) of AML patients stratified by protein abundance in ME22, with the most obvious prognostic discrimination used as the cutoff value.
#-----------------------------------------------------------------------------
#Step 1: Load data and and set parameters
#-----------------------------------------------------------------------------
AML_data<-readRDS("Input/AML_data.rds")
SNF<-readRDS("Input/SNF_group.rds")
PML<-SNF[SNF$WHO=="PML-RARA",]
SNF<-SNF[SNF$WHO!= PML$WHO,]
SNF<-SNF[SNF$SNF!= "SNF4",]
femData0 = AML_data$CorrectProteomics
femData<-femData0[,-c(1:2)]
rownames(femData)<-femData0$PG.ProteinGroups
femData.raw<-2^femData[,names(femData) %in% SNF$DIA_ID]
MetCox<-function (dat) {
metabolite_name <- names(dat)[3:ncol(dat)]
univ_formulas <- lapply(metabolite_name, function(x) stats::as.formula(paste("survival::Surv(time, status)~",
x)))
univ_models <- lapply(univ_formulas, function(x) {
survival::coxph(x, data = dat)
})
univ_results <- lapply(univ_models, function(x) {
x <- summary(x)
p.value <- signif(x$wald["pvalue"], digits = 2)
wald.test <- signif(x$wald["test"], digits = 2)
beta <- signif(x$coef[1], digits = 2)
HR <- signif(x$coef[2], digits = 2)
HR.confint.lower <- signif(x$conf.int[, "lower .95"],
2)
HR.confint.upper <- signif(x$conf.int[, "upper .95"],
2)
HR <- paste0(HR, " (", HR.confint.lower, "-", HR.confint.upper,
")")
name <- rownames(x$coefficients)
res <- c(name, beta, HR, wald.test, p.value)
names(res) <- c("name", "beta", "HR (95% CI for HR)",
"wald.test", "p.value")
return(res)
})
res <- t(as.data.frame(univ_results, check.names = FALSE))
result <- as.data.frame(res)
result <- result %>% tibble::as_tibble()
return(result)
}
vec<-apply(femData.raw,1,mean)
Q1 <- quantile(vec, 0.25)
Q3 <- quantile(vec, 0.75)
IQR_value <- Q3 - Q1
lower_threshold <- Q1 - 2 * IQR_value
upper_threshold <- Q3 + 2 * IQR_value
non.outliers <- vec[vec >= lower_threshold & vec <= upper_threshold ]
non.outliers.name<-names(non.outliers)
femData.raw<-femData.raw[row.names(femData.raw) %in% non.outliers.name,]
sd <- apply(femData.raw, 1, function(x) {sd(x)})
sd.top1000<-order(sd, decreasing = TRUE)[1:4000]
femData<-femData.raw[sd.top1000,]
femData<-log2(femData)
datExpr0 = as.data.frame(t(femData))
#-----------------------------------------------------------------------------
#Step 2: Weighted correlation network analysis
#-----------------------------------------------------------------------------
gsg = goodSamplesGenes(datExpr0, verbose = 3)
gsg$allOK
if (!gsg$allOK)
{
if (sum(!gsg$goodGenes)>0)
printFlush(paste("Removing genes:", paste(names(datExpr0)[!gsg$goodGenes], collapse = ", ")));
if (sum(!gsg$goodSamples)>0)
printFlush(paste("Removing samples:", paste(rownames(datExpr0)[!gsg$goodSamples], collapse = ", ")));
datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}
sampleTree = hclust(dist(datExpr0), method = "average")
cut=200
clust = cutreeStatic(sampleTree, cutHeight = cut, minSize = 10)
table(clust)
keepSamples = (clust==1)
datExpr = datExpr0[keepSamples, ]
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
traitData = AML_data$sampleinfo
SNF$value<-1
SNF.wide<-dcast(SNF,DIA_ID ~ SNF, value.var = "value")
traitData<-merge(SNF.wide,traitData,by="DIA_ID",all=F)
dim(traitData)
names(traitData)
allTraits = traitData[, c( "Sex", "Age" ,"BM_blasts" ,"WBC" , "HGB" , "PLT", "ELN_risk_2017" , "biCEBPA", "EFS_status" , "OS_status" , "SNF1" , "SNF2", "SNF3", "SNF5" , "SNF6" , "SNF7","SNF8" )]
allTraits$Sex<-gsub("M","1",allTraits$Sex)
allTraits$Sex<-gsub("F","0",allTraits$Sex)
allTraits<-data.frame(apply(allTraits,2,function (x){as.numeric(x)}))
allTraits[is.na(allTraits)]<-0
row.names(allTraits)<-traitData$DIA_ID
dim(allTraits)
names(allTraits)
Samples = rownames(datExpr)
traitRows = match(Samples,row.names(allTraits))
datTraits = allTraits[traitRows, ]
collectGarbage()
sampleTree2 = hclust(dist(datExpr), method = "average")
traitColors = numbers2colors(datTraits, signed = FALSE)
powers = c(c(1:20))
sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
sizeGrWindow(9, 5)
par(mfrow = c(1,2))
cex1 = 0.9
cor <- WGCNA::cor
net = blockwiseModules(datExpr, power =6,
TOMType = "unsigned", minModuleSize =20,
reassignThreshold = 0, mergeCutHeight =0.01,
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs = TRUE,
saveTOMFileBase = "AML_Proteomics",
verbose = 3)
moduleLabels = net$colors
moduleColors = labels2colors(net$colors)
MEs = net$MEs
geneTree = net$dendrograms[[1]]
net_score<-net$MEs
write.csv(net_score,"Output/Figure3/Net_score.csv")
#-----------------------------------------------------------------------------
#Step 3: Event-free survival stratified by ME22
#-----------------------------------------------------------------------------
traitData = AML_data$sampleinfo
KM_plot<- net_score %>%
dplyr::select(ME22) %>%
mutate(DIA_ID= row.names(.))
data<-merge(KM_plot,traitData[,c("DIA_ID","OS","OS_status","EFS","EFS_status")],by="DIA_ID",all=F)
library(pROC)
roc_curve <- roc(data$EFS_status, data$ME22)
optimal_cutoff_EFS <- coords(roc_curve, "best", ret = "threshold")
cutoff<-optimal_cutoff_EFS$threshold
data$group <- ifelse(data$ME22 > cutoff, "High", "Low")
fit <- survfit(Surv(EFS,EFS_status) ~ group, data)
EFS<-ggsurvplot(fit, data = data,
linetype = "strata",
pval = TRUE,
risk.table = T, tables.height = 0.3,
#tables.theme = theme_cleantable(),
xlim = c(0,1080),
break.x.by = 180,
ylab="Event-free survival",xlab = "Time (Days)",
surv.median.line = "hv",
font.x = c(14, "bold", "black"),
font.y = c(14, "bold", "black"),
font.tickslab = c(12, "plain", "black"),
palette = c("#E41A1C","#377EB8"),
legend.labs =c("High","Low"))
pdf("Output/Figure3/Figure3B_1.pdf", width = 8, height = 6)
print(EFS)
dev.off()
#-----------------------------------------------------------------------------
#Step 3: Overall survival stratified by ME22
#-----------------------------------------------------------------------------
fit <- survfit(Surv(OS,OS_status) ~ group, data)
OS<-ggsurvplot (fit, data = data,
linetype = "strata",
pval = TRUE,
risk.table = T, tables.height = 0.3,
#tables.theme = theme_cleantable(),
xlim = c(0,1080),
break.x.by = 180,
ylab="Overall survival",xlab = "Time (Days)",
surv.median.line = "hv",
font.x = c(14, "bold", "black"),
font.y = c(14, "bold", "black"),
font.tickslab = c(12, "plain", "black"),
palette = c("#E41A1C","#377EB8"),
legend.labs =c("High","Low"))
pdf("Output/Figure3/Figure3B_2.pdf", width = 8, height = 6)
print(OS)
dev.off()
4.4 (D) Pathway enrichment analysis
Pathway enrichment analysis of ME22-derived proteins between the combination of S2, S3, and S7 and residual AML patients using biological process (BP), cellular component (CC), and molecular function (MF) in Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG).
library(clusterProfiler)
library(org.Hs.eg.db)
library(ggplot2)
library(GOplot)
library(ggrepel)
#-----------------------------------------------------------------------------
#Step 1: Load data and and set parameters
#-----------------------------------------------------------------------------
limma_diff<-
function (dat, group)
{ sample_info<-data.frame(name= colnames(dat),group=group)
design <- stats::model.matrix(~0 + as.factor(group), data = sample_info)
colnames(design) <- stringr::str_replace_all(colnames(design),
stringr::fixed("as.factor(group)"), "")
fit <- limma::lmFit(dat, design)
contrast <- limma::makeContrasts(P_N = subtype - others, levels = design)
fits <- limma::contrasts.fit(fit, contrast)
ebFit <- limma::eBayes(fits)
deg_sig_list <- limma::topTable(ebFit, coef = 1, adjust.method = "fdr",
number = Inf)
deg <- deg_sig_list[which(!is.na(deg_sig_list$adj.P.Val)),
]
deg$logP <- -log10(deg$adj.P.Val)
deg$name <- rownames(deg)
return(tibble::as_tibble(deg))
}
prot.group<-readRDS("Input/WGCNA_result.rds")
info<-AML_data$sampleinfo
SNF<- readRDS("Input/SNF_group.rds")
prot0<-AML_data$CorrectProteomics
prot1<-prot0[,names(prot0) %in% SNF$DIA_ID]
row.names(prot1)<-prot0$PG.ProteinGroups
prot<-prot1[!grepl(";",row.names(prot1)),]
prot.group<-moduleColors
names(prot.group)<-names(net$colors)
prot.group<-data.frame(prot.group)
names(prot.group)<-"WGCNA_Groups"
prot.group$prot<-row.names(prot.group)
prots <- prot.group[prot.group$WGCNA_Groups == "darkgreen", ]
prots.name <- c(as.matrix(prots$prot))
protID = bitr(
prots.name,
fromType = "UNIPROT",
toType = c("ENSEMBL", "ENTREZID", "SYMBOL"),
OrgDb = "org.Hs.eg.db"
)
go_MF <-
enrichGO(
protID$SYMBOL,
OrgDb = org.Hs.eg.db,
ont = 'MF',
pAdjustMethod = 'BH',
pvalueCutoff = 0.05,
qvalueCutoff = 0.2,
keyType = 'SYMBOL'
)
go_CC <-
enrichGO(
protID$SYMBOL,
OrgDb = org.Hs.eg.db,
ont = 'CC',
pAdjustMethod = 'BH',
pvalueCutoff = 0.05,
qvalueCutoff = 0.2,
keyType = 'SYMBOL'
)
go_BP <-
enrichGO(
protID$SYMBOL,
OrgDb = org.Hs.eg.db,
ont = 'BP',
pAdjustMethod = 'BH',
pvalueCutoff = 0.05,
qvalueCutoff = 0.2,
keyType = 'SYMBOL'
)
Kegg <- enrichKEGG(
protID$ENTREZID,
organism = "hsa",
keyType = "kegg",
pvalueCutoff = 0.05,
pAdjustMethod = "BH",
minGSSize = 10,
maxGSSize = 500,
qvalueCutoff = 0.2,
use_internal_data = FALSE
)
bubble.go.MF <- data.frame(go_MF) %>%
top_n(n = 10, wt = -p.adjust)
bubble.go.MF$category <- "MF"
bubble.go.BP <- data.frame(go_BP) %>%
top_n(n = 10, wt = -p.adjust)
bubble.go.BP$category <- "BP"
bubble.go.CC <- data.frame(go_CC) %>%
top_n(n = 10, wt = -p.adjust)
bubble.go.CC$category <- "CC"
if (nrow(data.frame(Kegg)) > 0) {
bubble.KEGG <- data.frame(Kegg) %>%
top_n(n = 10, wt = -p.adjust)
bubble.KEGG$category <- "KEGG"
library(stringr)
bubble.KEGG.name <-
str_split_fixed(bubble.KEGG$geneID, "/", nrow(protID))
for (r in 1:nrow(bubble.KEGG)) {
for (c in 1:nrow(protID)) {
name.tmp <- bubble.KEGG.name[r, c]
name.tmp2 <- unique(protID[protID$ENTREZID == name.tmp, 4])
if (length(name.tmp2) == 0) {
name.tmp2 <- NA
} else{
name.tmp2 <- name.tmp2
}
bubble.KEGG.name[r, c] <- name.tmp2
}
}
bubble.KEGG.name.1 <-
data.frame(cbind(bubble.KEGG.name, new_col = apply(bubble.KEGG.name, 1, function(row) {
paste(row[!is.na(row)], collapse = ",")
})))
bubble.KEGG$geneID <- bubble.KEGG.name.1$new_col
} else {
bubble.KEGG <- data.frame(Kegg)
}
bubble.KEGG1 <- bubble.KEGG[, names(bubble.go.BP)]
bubble <- rbind(bubble.go.MF, bubble.go.BP, bubble.go.CC, bubble.KEGG1)
bubble.cir <-
data.frame(bubble$category,
bubble$ID,
bubble$Description,
bubble$geneID,
bubble$p.adjust)
names(bubble.cir) <-
c("Category", "ID" , "Term" , "Genes", "adj_pval")
bubble.cir$Genes <- gsub("/", ",", bubble.cir$Genes)
prot.mat <- prot
prot.diff.mat <-
prot.mat[row.names(prot.mat) %in% prots$prot, -ncol(prot.mat)]
prot.diff.mat.1 <- data.frame(t(prot.diff.mat))
prot.diff.mat.1$DIA_ID <- row.names(prot.diff.mat.1)
prot.diff.mat.2 <- merge(SNF, prot.diff.mat.1, by = "DIA_ID", all =F)
type <- prot.diff.mat.2$SNF
type[type %in% c("SNF2", "SNF7", "SNF3")] <- "subtype"
type[type != "subtype"] <- "others"
mat0 <- prot.diff.mat.2[, -c(1:15)]
row.names(mat0) <- prot.diff.mat.2$DIA_ID
mat <- t(mat0)
diff_meta0 <- limma_diff(mat, type)
diff_meta0$UNIPROT <- diff_meta0$name
diff_meta1 <- merge(protID[, c(1, 4)], diff_meta0, by = "UNIPROT")
diff_meta2 <- diff_meta1[, -1]
names(diff_meta2)[1] <- "ID"
circ <- circle_dat(bubble.cir, diff_meta2)
circ2 <- unique(circ[, -c(5, 6)])
#-----------------------------------------------------------------------------
#Step 2: Bubble plot
#-----------------------------------------------------------------------------
p <- ggplot(circ2, aes(x = zscore, y = -log10(adj_pval))) +
geom_point(aes(size = count, color = category), alpha = 0.6) +
scale_color_manual(values = c('palegreen4', 'orangered', 'skyblue', 'pink')) +
scale_size(range = c(1, 15)) +
theme_bw() +
geom_text_repel(
data = circ2[-log10(circ2$adj_pval) > 5, ],
aes(label = term),
size = 3,
segment.color = "black",
show.legend = FALSE
)
ggsave(
paste0("Output/Figure3/Figure3D.pdf") ,
plot = p,
device = NULL,
path = NULL,
width = 10,
height = 6
)