3 Figure 2
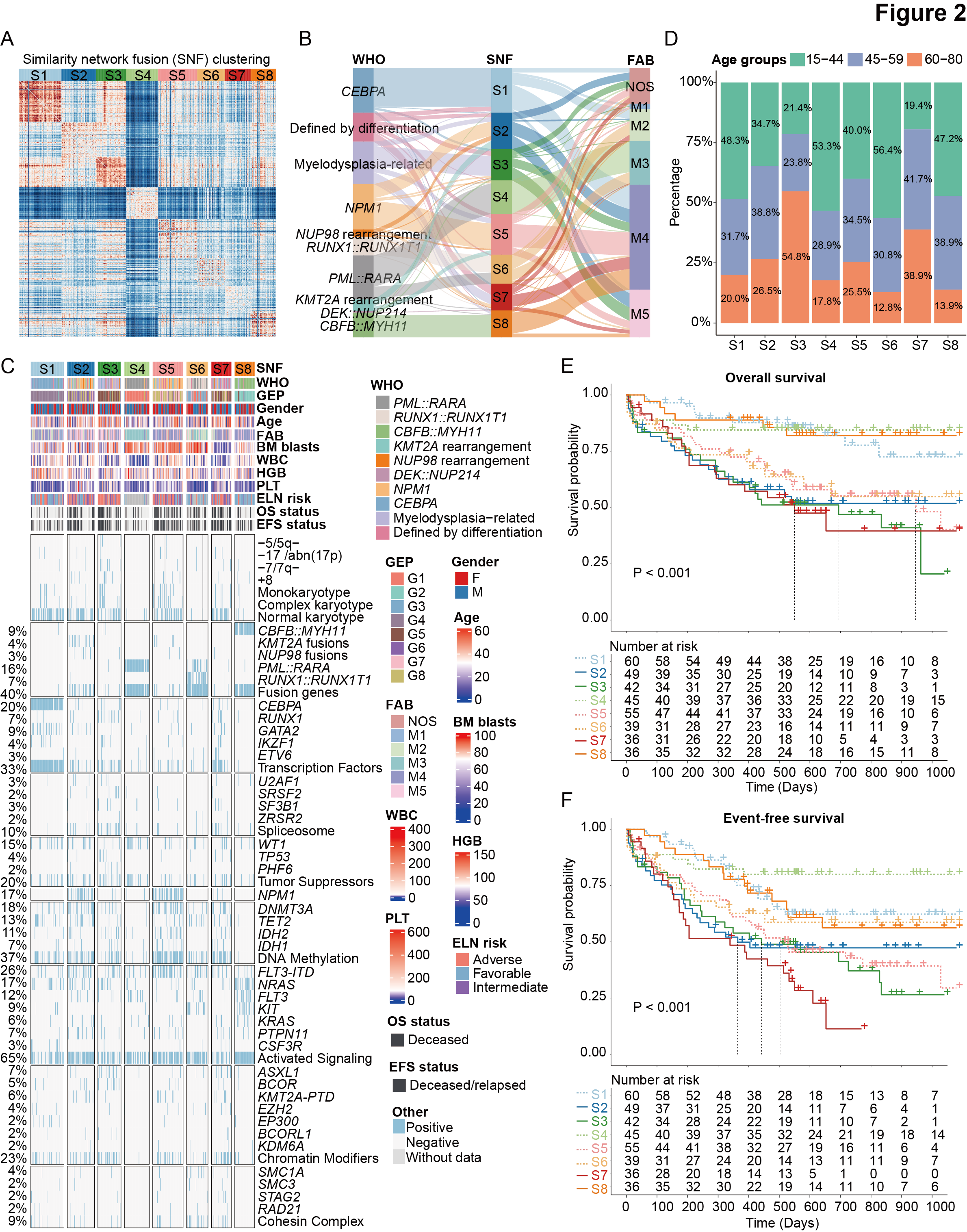
Figure 2. Proteomic subtypes of AML identified by similarity network clustering.
A. The discrimination of eight proteomic subtypes (S1-S8) based on the similarity network clustering (SNF) method.
B. Sankey plot indicating the relationship between the established proteomic subtypes and entities defined by the WHO and FAB classification.
C. Heatmap of clinical features, cytogenetic groups, and recurrent gene fusions and mutations in AML, which are classified into diverse functional groups (Ley TJ et al., N Engl J Med. 2013). Each column represents a patient and is arranged according to the proteomic clusters through S1-S8. “FLT3” refers to other FLT3 mutations except for FLT3-ITD, including FLT3-TKD mutations and mutations at other sites of this gene.
D. The proportional distribution of age groups in S1-S8 subtypes.
E-F. Kaplan-Meier curves for overall survival (E) and event-free survival (F) of AML patients stratified by the eight proteomic subtypes.
3.1 (A) SNF-based Proteomic subtypes
The discrimination of eight proteomic subtypes (S1-S8) based on similarity network clustering (SNF) method.
rm(list=ls())
library(dplyr)
library(SNFtool)
library(colorRamp2)
#-----------------------------------------------------------------------------
#Step 1: Load data and and set parameters
#-----------------------------------------------------------------------------
AML_data<-readRDS("Input/AML_data.rds")
prot0<-AML_data$CorrectProteomics
sampleinfo<-AML_data$sampleinfo
prot<-prot0[,-c(1:2)]
rownames(prot)<-prot0$PG.ProteinGroups
vec<-apply(prot,2,sum)
mean_value <- mean(vec)
std_deviation <- sd(vec)
##delete outlier
threshold <- 2* std_deviation
outliers <- vec[( mean_value-vec) > threshold]
outliers.name<-names(outliers)
prot<-prot[,!names(prot) %in% outliers.name]
##select best parameters for SNF
# ec.df<-data.frame()
# for (protnum in seq(1000,5000,500)) {
# sd <- apply(prot, 1, function(x) {sd(x)})
# sd.top1000<-order(sd, decreasing = TRUE)[1:protnum]
# cv <- apply(prot, 1, function(x) {sd(x)/mean(x)})
# cv.top1000<-order(cv, decreasing = TRUE)[1:1000]
# var <- apply(prot, 1, function(x) { var(x)})
# var.top1000<-order(var, decreasing = TRUE)[1:1000]
# prot.var<-prot[var.top1000,]
# prot.cv<-prot[cv.top1000,]
# prot.sd<-prot[sd.top1000,]
# protSNF = standardNormalization(t(prot.sd))
# for (K in seq(10,30,5)) {
# for (alpha in c(0.3,0.5,0.7)) {
# Dist1 = (dist2(as.matrix(protSNF),as.matrix(protSNF)))^(1/2)
# W1 = affinityMatrix(Dist1, K = K,alpha)
# ec <- estimateNumberOfClustersGivenGraph(W1, NUMC=5:15)
# ec.df1<-data.frame(protnum,K,alpha,ec)
# names(ec.df1)<-c("protnum","k","alpha",names(ec))
# ec.df<-rbind(ec.df1,ec.df)
# }
# }
# }
#-------------------------------------------------------------------------------
# Step 2: SNF of AML
#-------------------------------------------------------------------------------
protnum<-2000
K<-30
alpha<-0.7
clusterNum<-8
sd <- apply(prot, 1, function(x) {sd(x)})
sd.top.n<-order(sd, decreasing = TRUE)[1:protnum]
prot.sd<-prot[sd.top.n,]
protSNF = standardNormalization(t(prot.sd))
Dist1 = (SNFtool::dist2(as.matrix(protSNF), as.matrix(protSNF)))^(1/2)
W = affinityMatrix(Dist1, K=K,alpha )
Clusters = spectralClustering(W,K=clusterNum)
Clusters<-paste0("SNF",Clusters)
clusterDF<-data.frame(row.names (W),Clusters)
names(clusterDF)<-c("DIA_ID","SNF")
#------------------------------------------------------------------------------
# Step 3: Heatmap
#------------------------------------------------------------------------------
W1<-data.frame(W)
W1$SNF<-Clusters
annotation_col <-data.frame(
SNF=factor(clusterDF$SNF),
row.names = clusterDF$DIA_ID
)
order<-annotation_col%>%
arrange(SNF )
SNF_color <- c("#A6CEE3", "#1F78B4", "#33A02C","#B2DF8A" ,"#FB9A99", "#FDBF6F","#E31A1C", "#FF7F00" )
names(SNF_color) <- paste0("SNF", seq(1, clusterNum, 1))
ann_colors <- list(SNF=SNF_color)
colors<- c("#053061", "#2166AC", "#4393C3" ,"#92C5DE", "#D1E5F0" ,"#F7F7F7", "#FDDBC7", "#F4A582", "#D6604D", "#B2182B","#67001F")
color_series <- colorRampPalette(colors)(10000)
W2<-W1[order(W1$SNF),order(W1$SNF)]
W2[W2>0.01]<-NA
W2[W2>0.0048]<-0.0048
W2[W2< 0.0018]<- 0.0018
heatmap <- pheatmap::pheatmap(
W2,
annotation_col = annotation_col,
annotation_colors = ann_colors,
color = color_series,
fontsize_col = 1,
cluster_rows = F,
cluster_cols = F,
show_rownames = F,
show_colnames = T,
fontsize = 10,
cellwidth = 1,
cellheight = 1,
filename ="Output/Figure2/Figure2A.pdf",width = 10,height = 15)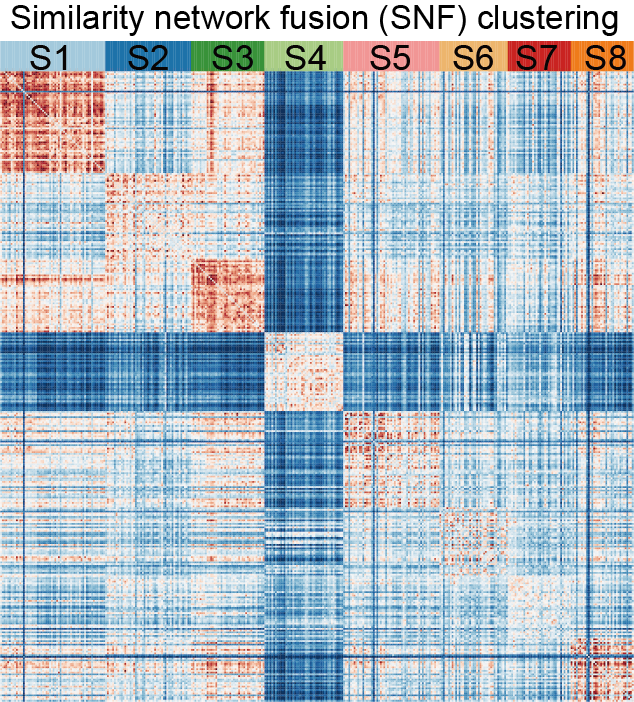
3.2 (C) Characterization of proteomic subtypes
Heatmap of clinical features, cytogenetic groups, and recurrent gene fusions and mutations in AML, which are classified into diverse functional groups. Each column represents a patient and is arranged according to the proteomic clusters through S1-S8.
#-----------------------------------------------------------------------------
#Step 1: Load data and and set parameters
#-----------------------------------------------------------------------------
source("Input/src/heatmap_new.R")
mutation.data1<-readRDS("Input/mutation.all.data.rds")
mutation.data2<-AML_data$TES_WES
mutation.data3<-mutation.data2[63:104,]
mutation.data4<-mutation.data3[,-1]
row.names(mutation.data4)<-mutation.data3$X
mutation.data5<-data.frame(t(mutation.data4),check.names=F)
mutation.data5$DIA_ID<-row.names(mutation.data5)
mutation.data6<-merge(mutation.data1,mutation.data5,by="DIA_ID",all=F)
mutation.all.data<-mutation.data6
annotation_col1 <-data.frame(
"-5/5q-" = factor(sampleinfo$Minus5_5q),
"-17 /abn(17p)" = factor(sampleinfo$Minus17_abn17p),
"-7/7q-" = factor(sampleinfo$Minus7_7q),
"+8" = factor(sampleinfo$Trisomy8),
Monokaryotype = factor(sampleinfo$Monokaryotype),
Complex_karyotype = factor(sampleinfo$Complex_karyotype),
Normal_karyotype = factor(sampleinfo$Normal_karyotype),
row.names = sampleinfo$DIA_ID,check.names = F
)
annotation_col1$DIA_ID<-row.names(annotation_col1)
mutation.all.data.all<-merge(annotation_col1,mutation.all.data,by="DIA_ID")
heatmap<-mutation.all.data.all[,-1]
row.names(heatmap)<-mutation.all.data$DIA_ID
heatmap1<-data.frame(t(heatmap))
heatmap2<-heatmap1[,names(W2)]
heatmap3<-data.frame(apply(heatmap2,2,function (x){as.numeric(x)}))
rownames(heatmap3)<-rownames(heatmap2)
WHO_order<-c(
"PML-RARA","RUNX1::RUNX1T1","CBFB::MYH11","KMT2A rearrangement","NUP98 rearrangement", "DEK::NUP214","NPM1","CEBPA","Myelodysplasia-related", "defined by differentiation"
)
annotation_col <-data.frame(
ELN_risk_2017 = factor(sampleinfo$ELN_risk_2017),
PLT = factor(sampleinfo$PLT),
HGB = factor(sampleinfo$HGB),
WBC = factor(sampleinfo$WBC),
BM_blasts = factor(sampleinfo$BM_blasts),
Diagnosis = factor(sampleinfo$Diagnosis),
Age = factor(sampleinfo$Age),
Sex= factor(sampleinfo$Sex),
WHO = factor(sampleinfo$WHO_classification_2022,levels = WHO_order),GEP=sampleinfo$tag2,
EFS_status = factor(sampleinfo$EFS_status),
OS_status = factor(sampleinfo$OS_status),
row.names = sampleinfo$DIA_ID,
check.names = F
)
annotation_col$ELN_risk_2017[annotation_col$ELN_risk_2017=="NA"]<-NA
annotation_col.tmp2<-apply(annotation_col[1:5],2,function (x) {as.numeric(x)})
annotation_col<-data.frame(annotation_col.tmp2,annotation_col[,c(6:12)],check.names = F)
SNF<-clusterDF
annotation_col$DIA_ID<-row.names(annotation_col)
annotation_col_SNF<-merge(annotation_col,SNF,by="DIA_ID",all=F)
annotation_col<-annotation_col_SNF[,c("SNF" ,"WHO" ,"GEP","Sex","Age","Diagnosis","BM_blasts","WBC" ,"HGB" ,"PLT" ,"ELN_risk_2017","OS_status","EFS_status" )]
names(annotation_col)<-c("SNF" ,"WHO" ,"GEP","Gender","Age","Diagnosis","BM_blasts","WBC" ,"HGB" ,"PLT" ,"ELN_risk_2017","OS_status","EFS_status" )
annotation_col$ELN_risk_2017<-gsub("1","Favorable",annotation_col$ELN_risk_2017)
annotation_col$ELN_risk_2017<-gsub("2","Intermediate",annotation_col$ELN_risk_2017)
annotation_col$ELN_risk_2017<-gsub("3","Adverse",annotation_col$ELN_risk_2017)
row.names(annotation_col)<-annotation_col_SNF$DIA_ID
annotation_col$Age<-as.numeric(annotation_col$Age)
annotation_col<-annotation_col[names(W2),]
#-----------------------------------------------------------------------------
#Step 2: Heatmap
#-----------------------------------------------------------------------------
SNF_color <- SNF_color
names(SNF_color) <- paste0("SNF", seq(1, 8, 1))
ELN_risk_2017_color <- c("#80b1d3", "#9467bd", "#fb8072")
names(ELN_risk_2017_color) <- c("Favorable", "Intermediate", "Adverse")
WHO_color<- c("#999999" , "#e7dad2" , "#96c37d", "#8ecfc9","#FF7F00", "#c497b2","#ffbe7a", "#82b0d2" ,"#beb8dc", "#e984a2")
status_color = c("1" = "#434348","0" = "#eeeeee","NA" = "white")
status_color = c("1" = "#434348","0" = "#eeeeee","NA" = "white")
names(WHO_color) <-WHO_order
GEP_color <- c("#fb8072","#8dd3c7","#80b1d3","#998199","#8c564b","#9467bd","pink","lightgoldenrod3")
names(GEP_color) <- paste0("G", seq(1, 8, 1))
Diagnosis_color <- c("#DF9E9B","#99BADF","#D8E7CA","#99CDCE","#999ACD","#FFD0E9")
names(Diagnosis_color) <- c("AML","M1","M2","M3","M4","M5")
Gender_color<- c("#377EB8", "#E41A1C")
names(Gender_color) <- c("M", "F")
numbers<-c("#92C5DE", "#D1E5F0" ,"#F7F7F7", "#FDDBC7", "#F4A582")
number_color<-colorRampPalette(numbers)(1000)
Age_color = colorRamp2(c(0, 50, 100), c("blue", "white", "red"))
WBC_color = colorRamp2(c(0, 10, 300), c("blue", "white", "red"))
HGB_color = colorRamp2(c(0, 60, 150), c("blue", "white", "red"))
PLT_color = colorRamp2(c(0, 50, 400), c("blue", "white", "red"))
BM_color = colorRamp2(c(0, 70, 100), c("blue", "white", "red"))
Age_colors = Age_color(seq(0, 100, length.out = 100))
WBC_colors = WBC_color(seq(0, 300, length.out = 100))
HGB_colors = HGB_color(seq(0, 150, length.out = 100))
PLT_colors = PLT_color(seq(0, 400, length.out = 100))
BM_colors = BM_color(seq(0, 100, length.out = 100))
ann_colors <- list(Diagnosis = Diagnosis_color,WHO=WHO_color,Gender=Gender_color,SNF=SNF_color,Age=Age_colors,GEP=GEP_color,BM_blasts=BM_colors,WBC=WBC_colors,HGB=HGB_colors,PLT=PLT_colors,ELN_risk_2017=ELN_risk_2017_color,OS_status=status_color,EFS_status=status_color)
colors<- c( "#F7F7F7", "#92C5DE")
heatmap3<-heatmap3[,names(W2)]
heatmap4<-data.frame(apply(heatmap3,2,function (x) {as.numeric(x)}))
row.names(heatmap4)<-rownames(heatmap3)
column_split <- c(annotation_col$SNF)
column_split<-gsub("SNF","",column_split)
column_split<-as.numeric(column_split)
row_split <- c(rep(1, 7),rep(2, 6),rep(3, 6),rep(4, 5),rep(5, 4),rep(6, 1),rep(7, 5),rep(8, 8),rep(9, 8),rep(10,5))
percentages<-apply(heatmap4, 1, function (x){sum(x,na.rm = T)/ncol(heatmap4)})
percentage_df <- data.frame(Percentage = paste0(percentages * 100, "%"),
Row = rownames(heatmap4))
heatmap <- heatmap_new(column_split = column_split,
row_split = row_split,
column_gap = unit(2, "mm"),
border_gp = gpar(col = "black", row = "black", lwd = 1),
as.matrix(heatmap4),
color = colors,
cluster_rows = F,
cluster_cols = F,
annotation_col = annotation_col[, ncol(annotation_col):1],
annotation_colors = ann_colors,
left_annotation = rowAnnotation(
Percentage = anno_text(percentage_df$Percentage,
location = unit(0, "npc"),
just = "right"),
annotation_name_gp = gpar(fontsize = 10),
annotation_width = unit(1.5, "cm")
)
)
pdf("Output/Figure2/Figure2C.pdf", width = 10, height = 20)
draw(heatmap)
dev.off()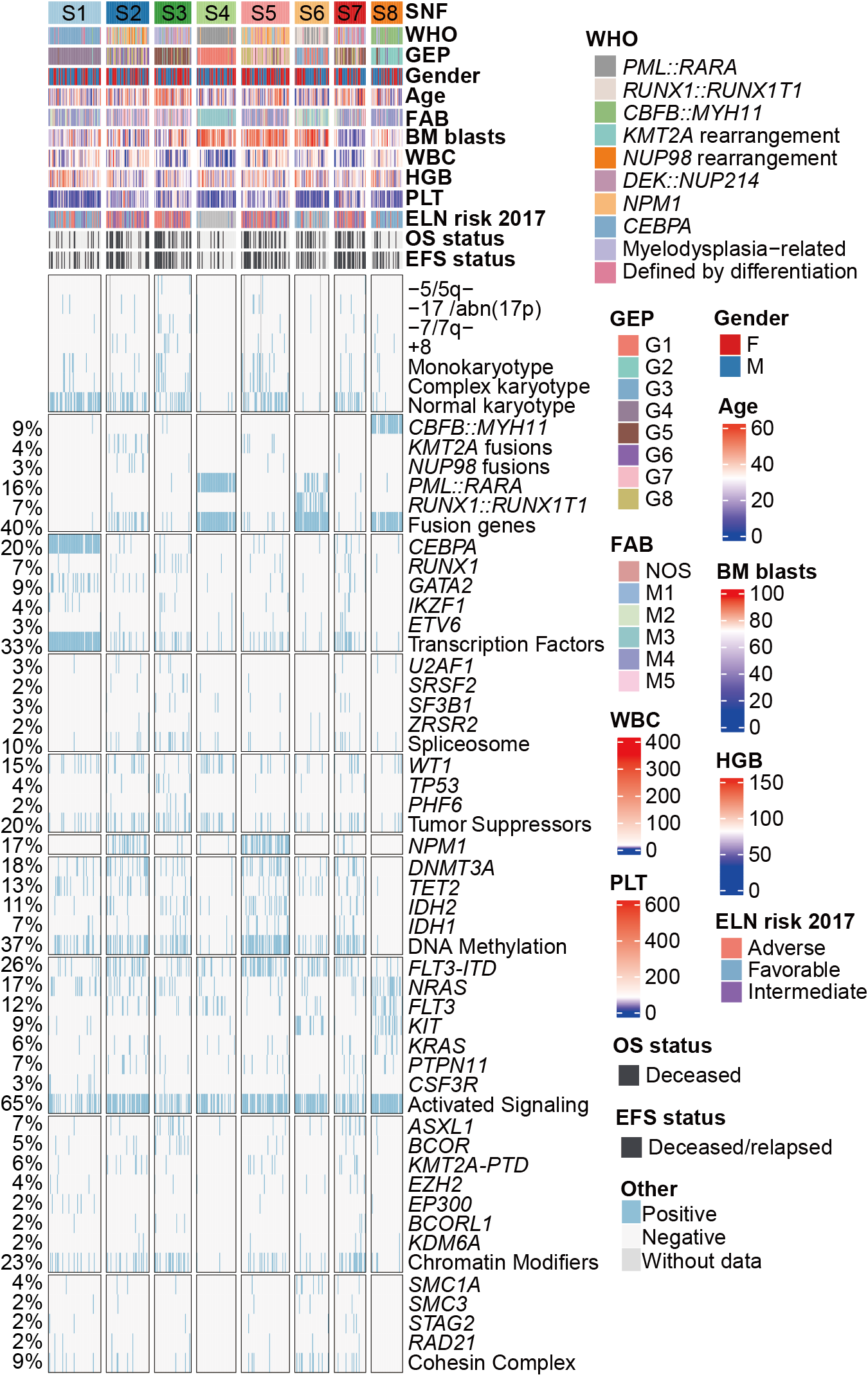
3.3 (E) Overall survival
Kaplan-Meier curves for overall survival of AML patients stratified by the eight proteomic subtypes.
library(survival)
library(survminer)
#-----------------------------------------------------------------------------
#Step 1: Load data and and set parameters
#-----------------------------------------------------------------------------
AML_data<-readRDS("Input/AML_data.rds")
prot0<-AML_data$CorrectProteomics
sampleinfo<-AML_data$sampleinfo
OS<-sampleinfo[,c("DIA_ID","OS","OS_status")]
clusterDF.km<-merge(clusterDF,OS,by="DIA_ID",all=F)
clusterDF.km$OS[clusterDF.km$OS > 1080 ] <-1080
clusterDF.km$OS_status <-ifelse(clusterDF.km$OS == 1080, 0, clusterDF.km$OS_status )
#-----------------------------------------------------------------------------
#Step 2: Overall survival analysis
#-----------------------------------------------------------------------------
survival.data<-clusterDF.km[,c("SNF","OS","OS_status")]
names(survival.data)<-c("Risk","Time","Event")
fit <- survfit(Surv(Time,Event) ~ Risk,
data = survival.data)
SNF_color <- c("#A6CEE3", "#1F78B4", "#33A02C","#B2DF8A" ,"#FB9A99", "#FDBF6F","#E31A1C", "#FF7F00" )
pdf("Output/Figure2/Figure2E.pdf", width = 20, height =20)
km_plot<- ggsurvplot(fit,
data = survival.data,
conf.int = F,
pval = TRUE,
surv.median.line = "hv",
risk.table = TRUE,
xlab = "Follow up time(d)",
legend = c(0.8,0.75),
legend.title = "",
linetype = c(8,1,1,8,8,8,1,1),
palette = SNF_color)
print(km_plot)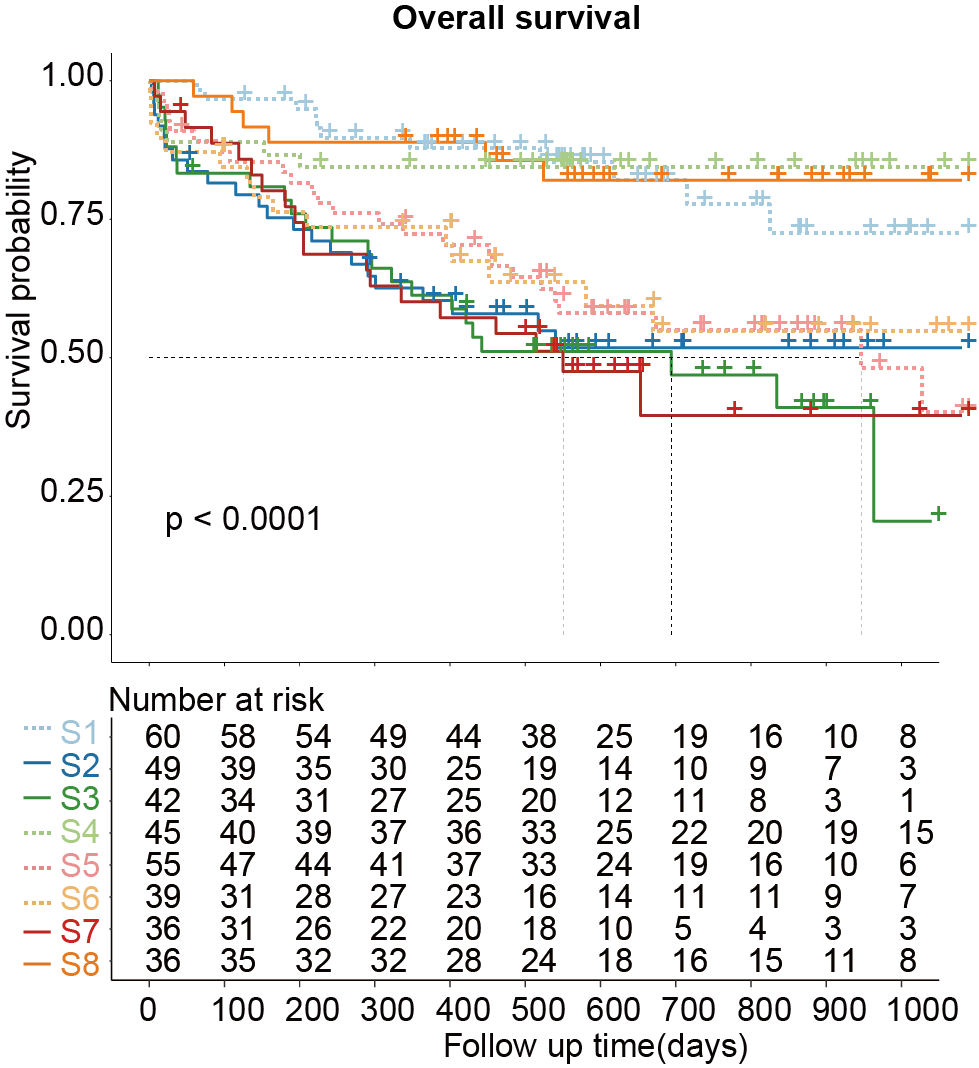
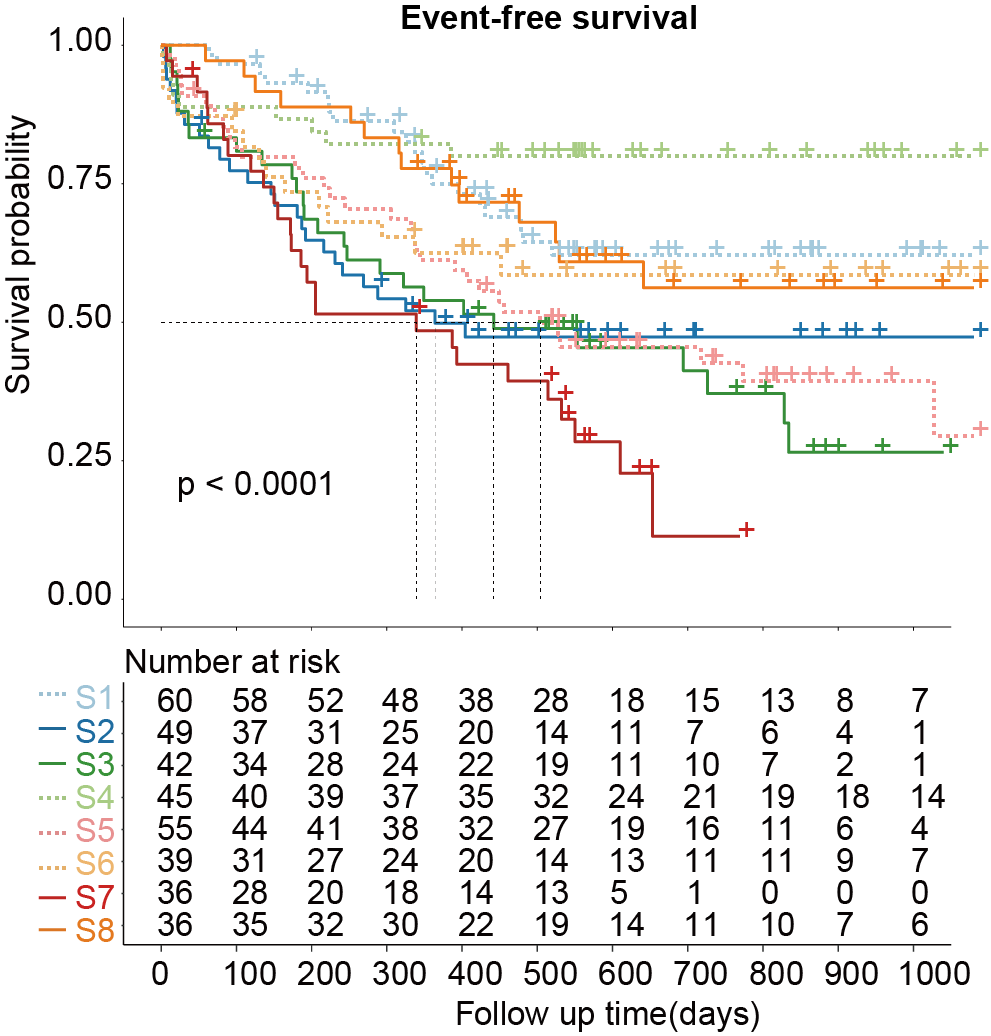
3.4 (F) Event-free survival
Kaplan-Meier curves for event-free survival of AML patients stratified by the eight proteomic subtypes.
library(survival)
library(survminer)
#-----------------------------------------------------------------------------
#Step 1: Load data and and set parameters
#-----------------------------------------------------------------------------
EFS<-sampleinfo[,c("DIA_ID","EFS","EFS_status")]
clusterDF.km<-merge(clusterDF,EFS,by="DIA_ID",all=F)
clusterDF.km$EFS[clusterDF.km$EFS > 1080 ] <-1080
clusterDF.km$EFS_status <-ifelse(clusterDF.km$EFS == 1080, 0, clusterDF.km$EFS_status )
#-----------------------------------------------------------------------------
#Step 2: Event-free survival analysis
#-----------------------------------------------------------------------------
survival.data<-clusterDF.km[,c("SNF","EFS","EFS_status")]
names(survival.data)<-c("Risk","Time","Event")
fit <- survfit(Surv(Time,Event) ~ Risk,
data = survival.data)
SNF_color <- c("#A6CEE3", "#1F78B4", "#33A02C","#B2DF8A" ,"#FB9A99", "#FDBF6F","#E31A1C", "#FF7F00" )
pdf("Output/Figure2/Figure2F.pdf", width = 20, height =20)
km_plot<- ggsurvplot(fit,
data = survival.data,
conf.int = F,
pval = TRUE,
surv.median.line = "hv",
risk.table = TRUE,
xlab = "Follow up time(d)",
legend = c(0.8,0.75),
legend.title = "",
break.x.by = 100,
linetype = c(8,1,1,8,8,8,1,1),
palette = SNF_color)
print(km_plot)