5 Figure 4
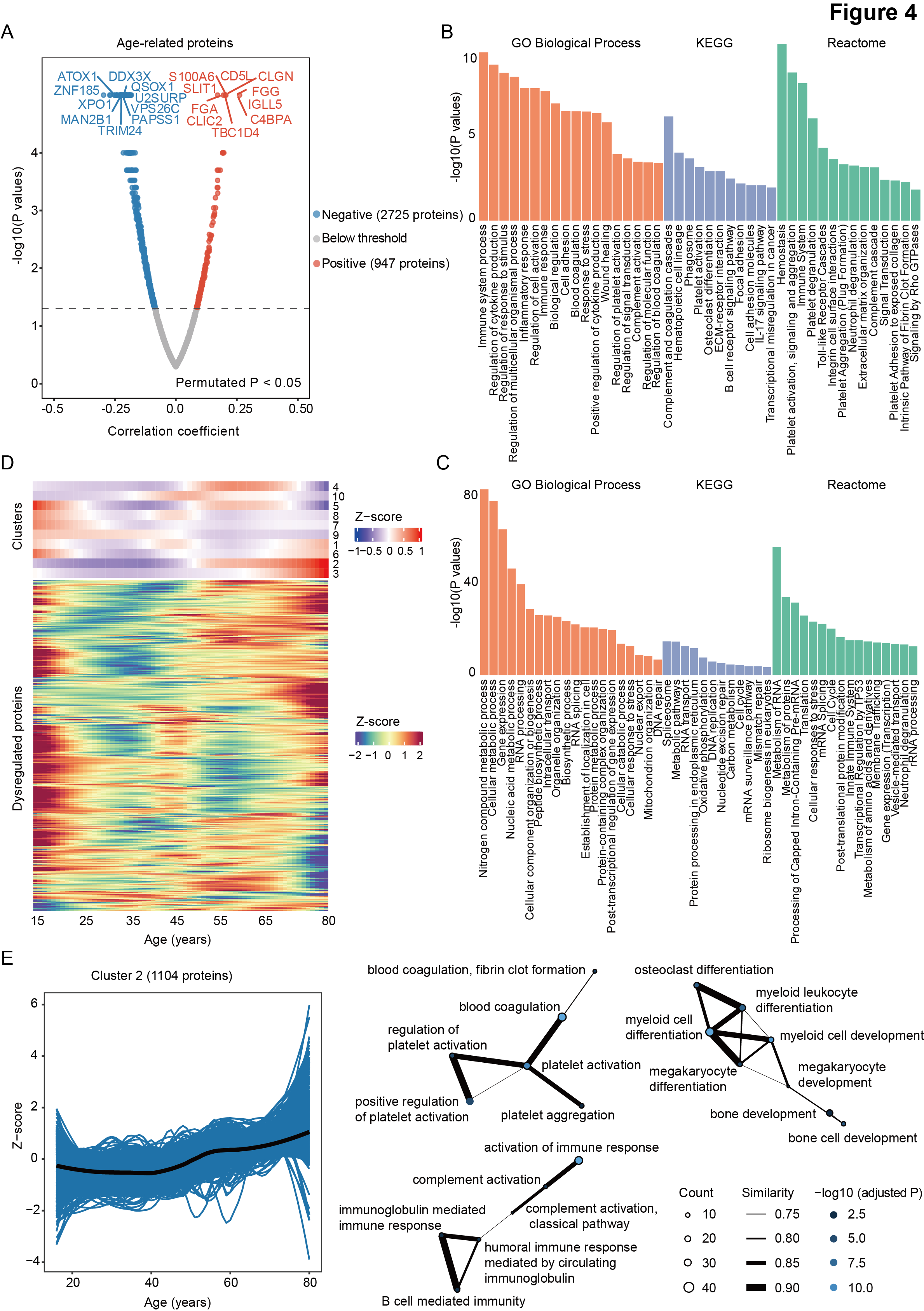
Figure 4. Changes in protein and phosphosite abundance associated with aging.
A Volcano plot of significantly changed proteins (permutated P < 0.05) during aging in AML, which were calculated using the Spearman correlation approach.
B-C Pathway enrichment analysis (GO Biological Process, KEGG, and Reactome) of proteins showing positive (B) and negative (C) correlation with age in all AML patients.
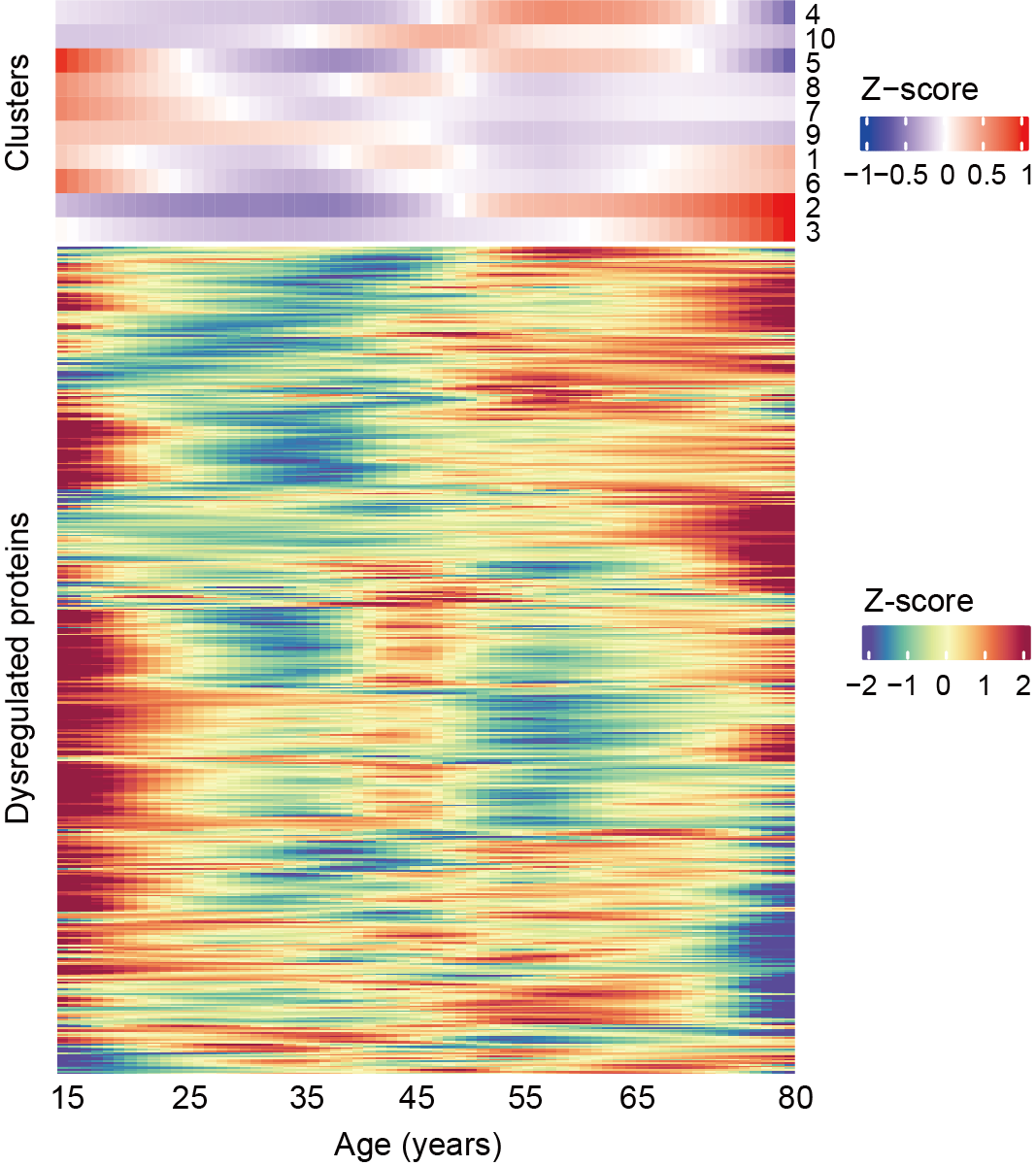
D. Heatmaps displaying the trajectories of 10 protein clusters (upper panel) and all proteins (lower panel) that change with age in AML.
E. Enriched GO pathways of proteins in cluster 2. The size of the circles represents the count of genes enriched in the GO term. The thickness of the edges denotes the semantic similarity between each pair of the GO terms. The color of the circles indicates the adjusted significance of enrichment. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
5.1 (A) Volcano plot
Volcano plot of significantly changed proteins during aging in AML.
rm(list=ls())
library(broom)
library(dplyr)
library(Mfuzz)
library(dplyr)
library(lubridate)
library(openxlsx)
library(readxl)
library(ggplot2)
library(ggrepel)
cor.pvalues_pearson<-read_xlsx("Input/cor.pvalues_pearson_protein_final (1).xlsx")
cor.pvalues_pearson[cor.pvalues_pearson$Log10.p_adjusted==Inf,]$Log10.p_adjusted<-5
color_values <- c(
"Pos" = "#E64B35",
"Neg" = "#3182BD",
"no" = "gray70"
)
top_n <- 10
top_points_pos <- cor.pvalues_pearson %>%
filter(Sig=="Pos") %>%
arrange(desc(Log10.p_adjusted)) %>%
head(top_n)
top_points_neg <- cor.pvalues_pearson %>%
filter(Sig=="Neg") %>%
arrange(desc(Log10.p_adjusted)) %>%
head(top_n)
ggplot(cor.pvalues_pearson, aes(x = cor, y = Log10.p_adjusted, color = Sig)) +
geom_point(alpha = 0.7, size = 2.5) +
# 添加参考线
geom_hline(yintercept = -log10(0.05), linetype = "dashed", color = "grey30") +
#geom_vline(xintercept = c(-0.5, 0.5), linetype = "dashed", color = "grey30") +
geom_text_repel(data = top_points_pos, aes(label = PG.Genes, color = Sig),
box.padding = 0.3, point.padding = 0.3,
segment.color = "#E64B35", # 设置短线颜色为 Pos 对应的颜色
segment.size = 0.5,
size = 4, max.overlaps = 50) +
geom_text_repel(data = top_points_neg, aes(label = PG.Genes, color = Sig),
box.padding = 0.3, point.padding = 0.3,
segment.color = "#3182BD", # 设置短线颜色为 Neg 对应的颜色
segment.size = 0.5,
size = 4, max.overlaps = 50) +
scale_color_manual(values = color_values) +
labs(
x = "Correlation Coefficient (r)",
y = "-log10(p-value)",
title = "Volcano Plot of Age-Gene Expression Correlation",
color = "Significance"
) +
# 主题美化
theme_classic(base_size = 12) +
theme(
plot.title = element_text(hjust = 0.5, face = "bold", size = 14),
legend.position = "right",
panel.border = element_rect(color = "black", fill = NA, linewidth = 0.5),
axis.text = element_text(color = "black"),
legend.background = element_blank()
) +
# 坐标轴范围调整(根据实际数据修改)
xlim(-0.5, 0.5) +
ylim(0, max(cor.pvalues_pearson$Log10.p_adjusted) + 0.2)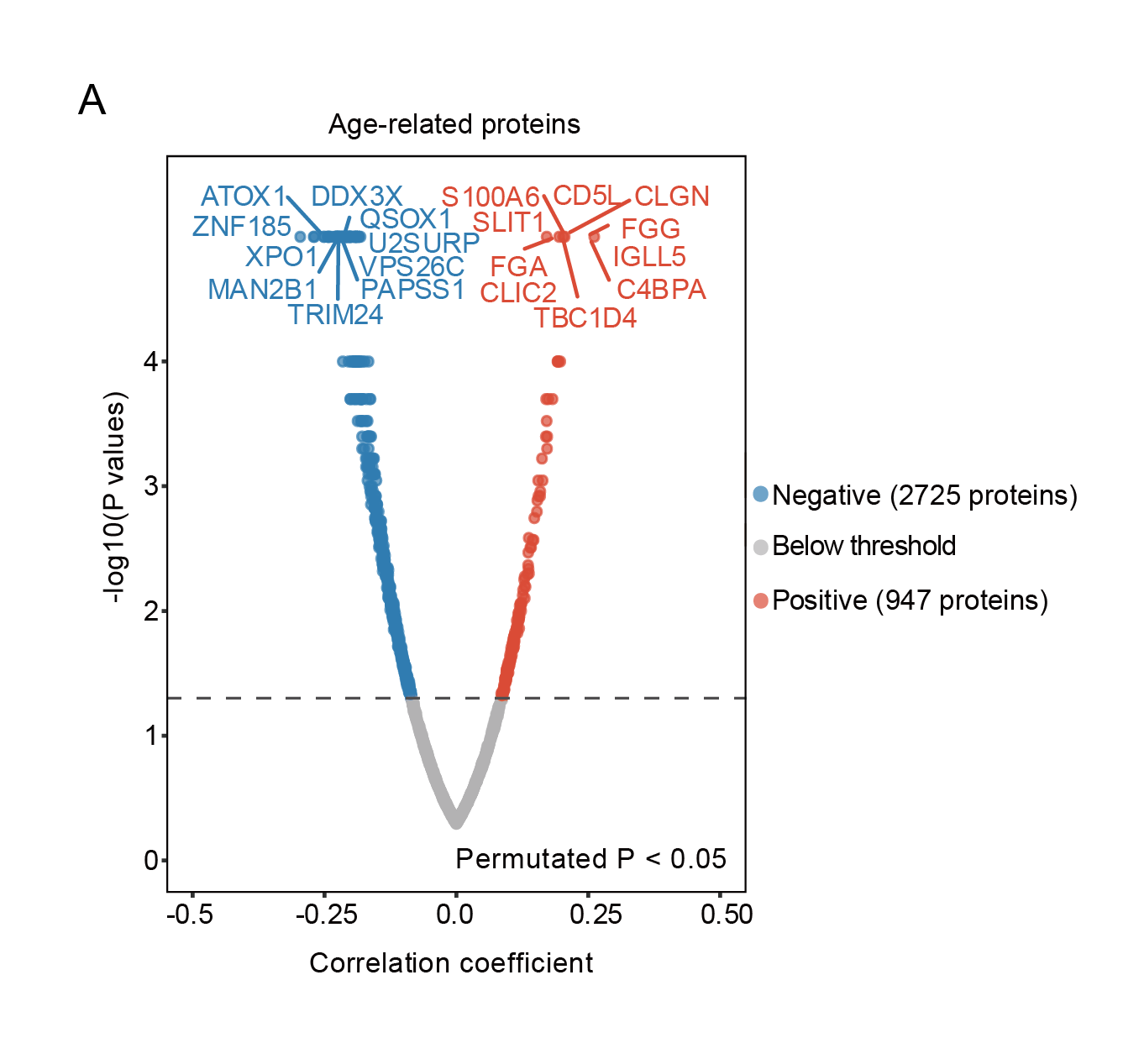
5.2 (B) bar plot
Bar plot (B) and negative (C) correlation with age in all AML patients.
rm(list=ls())
cor.pvalues_pearson<-read_xlsx("Input/cor.pvalues_pearson_protein_final (1).xlsx")
cor.pvalues_pearson_Pos <- cor.pvalues_pearson$PG.Genes[which(cor.pvalues_pearson$Sig == 'Pos')]
cor.pvalues_pearson_Pos <- unlist(strsplit(cor.pvalues_pearson_Pos, ";"))
cor.pvalues_pearson_selected.enrichment <- read_excel('Input/cor.pvalues_pearson_Pos.enrichment.selected.xls')
cor.pvalues_pearson_selected.enrichment <- cor.pvalues_pearson_selected.enrichment %>%
filter(selected...4 == 1)
unique(cor.pvalues_pearson_selected.enrichment$`#category`)
cor.pvalues_pearson_selected.enrichment$log10.fdr <- -log10(as.numeric(cor.pvalues_pearson_selected.enrichment$`false discovery rate`))
cor.pvalues_pearson_selected.enrichment <- cor.pvalues_pearson_selected.enrichment[, c('#category', 'term description', 'log10.fdr')]
df <- cor.pvalues_pearson_selected.enrichment
length(unique(cor.pvalues_pearson_selected.enrichment$`term description`))
df$color <- ifelse(df$`#category` == "GO Process", "#fc8d62",
ifelse(df$`#category` == "Reactome", "#66c2a5", "#8da0cb"))
df <- df %>%
mutate(`#category` = factor(`#category`, levels = c("GO Process", "KEGG","Reactome"))) %>%
arrange(`#category`, desc(log10.fdr))
df<-df[c(-14,-18),]
df$`term description` <- factor(df$`term description`, levels = unique(df$`term description`))
# 绘制条形图
options(repr.plot.width=15, repr.plot.height=10)
library(ggplot2)
ggplot(df, aes(x = `term description`, y = log10.fdr, fill = color)) +
geom_bar(stat = "identity") +
scale_fill_identity() +
#coord_flip() +
labs(x = "Enriched pathways", y = "-Log10(P values)") +
theme_minimal(base_size = 15) + # 增大字体
theme(legend.position = "top",
axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5, color = "black"), # x轴文字黑色
axis.text.y = element_text(color = "black"), # y轴文字黑色
axis.title.x = element_text(color = "black"), # x轴标题黑色
axis.title.y = element_text(color = "black"), # y轴标题黑色
panel.grid = element_blank()) + # 移除背景条纹
scale_y_continuous(breaks = seq(0, max(df$log10.fdr), by = 5)) # 可选:设置y轴刻度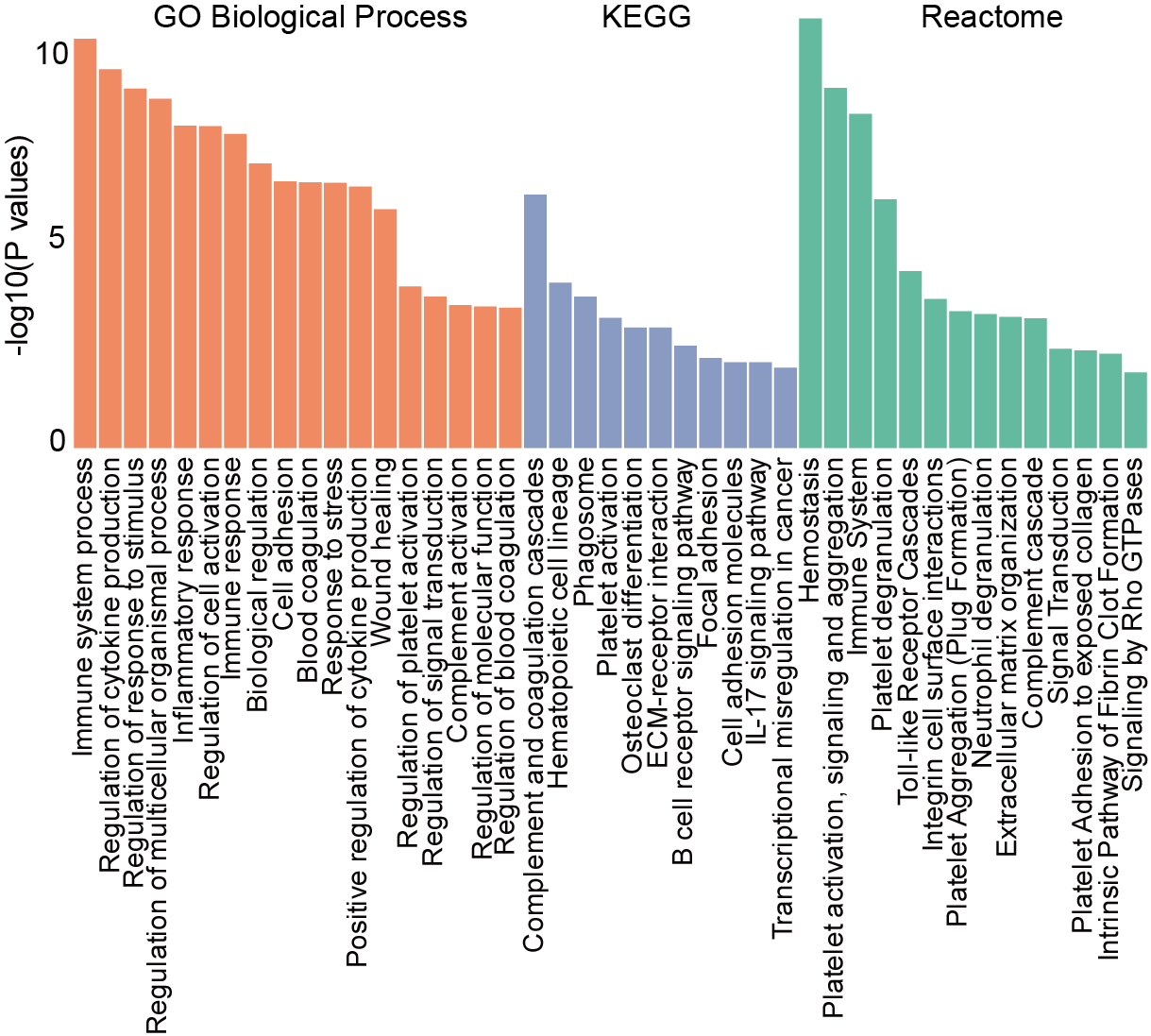
5.3 (D) Heatmaps
Heatmaps showing the trajectories of 10 protein clusters , as well as all nonlinear or linear changing proteins during aging in AML.
rm(list=ls())
library(tidyverse)
library(parallel)
library(ComplexHeatmap)
library(plyr)
library(openxlsx)
library(readxl)
library(Mfuzz)
library(dplyr)
library(ggplot2)
#-----------------------------------------------------------------------------
#Step 1: Loess
#-----------------------------------------------------------------------------
AML_data <- readRDS("Input/AML_data.rds")
clinical.features <-AML_data$sampleinfo
clinical.features$Age.Group <- as.character(cut(clinical.features$Age,
breaks = c(15, 44, 59, 80),
labels = c("15-44",
"45-59",
"60-80"),
right = TRUE,
include.lowest = TRUE
))
protein.abundance <- AML_data$CorrectProteomics
protein.meta <- protein.abundance[, c(1,2)]
rownames(protein.abundance) <- protein.abundance[,1]
protein.abundance <- protein.abundance[, -c(1,2)]
protein.abundance <- t(protein.abundance)
protein.abundance <- as.data.frame(protein.abundance)
protein.abundance$Age <- as.numeric(plyr::mapvalues(rownames(protein.abundance),
from=clinical.features$DIA_ID,
to=clinical.features$Age))
sample_info <- data.frame(sampleID=rownames(protein.abundance), Age = protein.abundance$Age)
variable_info <- protein.meta
colnames(variable_info) <- c('variable_id','PG.Genes')
new_expression_data<-readRDS("Input/loess.new_expression_data.RDS")
library(circlize)
temp_data <- new_expression_data
max(temp_data)
min(temp_data)
temp_data <- temp_data %>%
apply(1, function(x) {
x-mean(x)
}) %>%
as.data.frame()
temp_data<- temp_data %>%
scale() %>%
t()
col_fun = colorRamp2(seq(-2, 2, length.out = 11),
rev(RColorBrewer::brewer.pal(n = 11, name = "Spectral")))
custom_order <- c(2, 3, 4, 10, 1, 8, 7, 9, 6, 5)
cluster_info<-readRDS("Input/final_cluster_info2.0.RDS")
cluster_info$cluster <- factor(cluster_info$cluster, levels = custom_order)
ordered_data <- cluster_info[order(cluster_info$cluster), ]
result_vector <- ordered_data[[1]]
reordered_matrix <- temp_data[match(result_vector, rownames(temp_data)), ]
plot <-
Heatmap(
reordered_matrix ,
show_column_names = FALSE,
show_row_names = FALSE,
cluster_columns = FALSE,
cluster_rows = TRUE,
show_row_dend = TRUE,
col = col_fun,
border = TRUE
)
plot
#-----------------------------------------------------------------------------
#Step 2: Mfuzz
load("Input/Mfuzz.RData")
proteomics_cluster_info <-
final_cluster_info
dim(final_cluster_info)
temp <-
final_cluster_info %>%
dplyr::count(cluster) %>%
dplyr::filter(n > 1)
final_cluster_info <-
final_cluster_info %>%
dplyr::filter(cluster %in% temp$cluster)
proteomics_data <-
readr::read_delim(
"Input/temp_data.txt"
)
row_names <-
proteomics_data$...1[-1]
proteomics_data <-
proteomics_data[-1, ] %>%
tibble::column_to_rownames(var = "...1") %>%
purrr::map(function(x) {
as.numeric(x)
}) %>%
dplyr::bind_cols()
rownames(proteomics_data) <-
row_names
expression_data <- readRDS('Input/loess.new_expression_data.RDS')
proteomics_all_data <-
expression_data %>%
apply(1, function(x) {
(x - mean(x))
}) %>%
t() %>%
as.data.frame()
temp <-
proteomics_cluster_info %>%
dplyr::count(cluster) %>%
dplyr::filter(n > 1)
proteomics_cluster_info <-
proteomics_cluster_info %>%
dplyr::filter(cluster %in% temp$cluster)
proteomics_mean_data <-
unique(proteomics_cluster_info$cluster) %>%
purrr::map(function(idx) {
if (sum(proteomics_cluster_info$cluster == idx) == 1) {
return(NULL)
}
proteomics_data[proteomics_cluster_info$variable_id[proteomics_cluster_info$cluster == idx],] %>%
colMeans()
}) %>%
dplyr::bind_rows() %>%
as.data.frame()
rownames(proteomics_mean_data) <-
paste("proteomics", unique(proteomics_cluster_info$cluster), sep = "_")
proteomics_mean_all_data <-
unique(proteomics_cluster_info$cluster) %>%
purrr::map(function(idx) {
if (sum(proteomics_cluster_info$cluster == idx) == 1) {
return(NULL)
}
proteomics_all_data[proteomics_cluster_info$variable_id[proteomics_cluster_info$cluster == idx],] %>%
colMeans()
}) %>%
dplyr::bind_rows() %>%
as.data.frame()
rownames(proteomics_mean_all_data) <-
paste("proteomics",
unique(proteomics_cluster_info$cluster),
sep = "_")
temp_data <- proteomics_mean_data
temp_data_all <- proteomics_mean_all_data
col_fun = colorRamp2(c(-2, 0, 2),
c(
viridis::viridis(n = 3)[1],
viridis::viridis(n = 3)[2],
viridis::viridis(n = 3)[3]
))
library(circlize)
ha <- rowAnnotation(
class = rownames(temp_data) %>% stringr::str_replace("_[0-9]{1,2}", ""),
#col = list(class = omics_color),
na_col = "black",
border = TRUE,
gp = gpar(col = "black")
)
plot <-
Heatmap(
temp_data,
cluster_columns = FALSE,
border = TRUE,
column_names_rot = 45,
rect_gp = gpar(col = "black"),
col = col_fun,
name = "Z-score",
clustering_distance_columns = "euclidean",
clustering_method_columns = "ward.D2",
right_annotation = ha
)
ha <- rowAnnotation(
class = rownames(temp_data_all) %>% stringr::str_replace("_[0-9]{1,2}", ""),
#col = list(class = omics_color),
na_col = "black",
border = TRUE,
gp = gpar(col = "black")
)
col_fun <- colorRamp2(c(-1, 0, 1),
c("blue", "white", "red"))
plot <- Heatmap(
temp_data_all[c(
'proteomics_2',
'proteomics_3',
'proteomics_4',
'proteomics_10',
'proteomics_1',
'proteomics_8',
'proteomics_7',
'proteomics_9',
'proteomics_6',
'proteomics_5'
),],
cluster_columns = FALSE, # 不聚类列
cluster_rows = FALSE, # 聚类行
border = FALSE, # 不显示边框
column_names_rot = 0, # 列名旋转
show_column_names = TRUE, # 不显示列名
#rect_gp = gpar(col = "black"), # 边框颜色设置为黑色,但实际上不显示
col = col_fun,
name = "Z-score",
clustering_distance_columns = "euclidean",
clustering_method_columns = "complete",
right_annotation = ha
)
options(repr.plot.width=20, repr.plot.height=3)
plot