6.1 Number of mutation (boxplot)
# prepare data
df_nummut <- df_full %>% group_by(ID,gene) %>% summarize(nummut=sum(vaf>0)) %>% ungroup() %>% mutate(nummut=ifelse(is.na(nummut),0,nummut))
df_maxvaf <- df_full %>% group_by(ID,gene) %>% summarize(maxvaf=max(vaf)) %>% ungroup() %>% mutate(maxvaf=ifelse(is.na(maxvaf),0,maxvaf))
df_all <- df_input %>% inner_join(df_nummut, by=c('ID','gene')) %>% inner_join(df_maxvaf, by=c('ID','gene'))
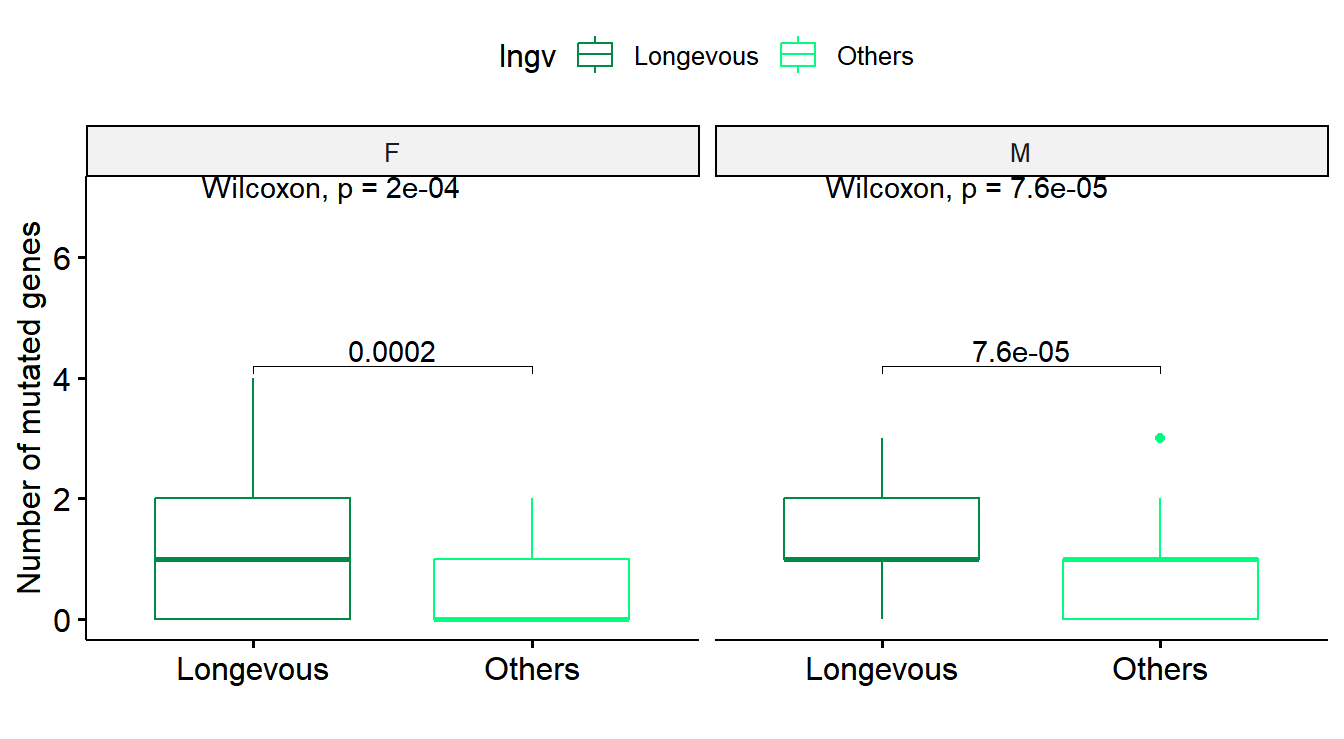
# Figure 2A
# mutated genes drawn separately by gender
df_tmp <- df_all %>% group_by(ID,Sex,Age,agrp,lngv) %>% summarise(numgene=sum(nummut>0)) %>% ungroup()
gp <- df_tmp %>% ggboxplot(x="lngv", y="numgene", color="lngv", palette=c("springgreen4","springgreen"))
gp <- gp + stat_compare_means(comparisons=list(c("Longevous", "Others")), method="wilcox.test") + stat_compare_means(label.y=7) + xlab("") + ylab("Number of mutated genes")
gp_box <- facet(gp, facet.by="Sex")
# saved into a file 'Boxplot_numgene.pdf'
file.path(outdir,"Boxplot_numgene.pdf") %>% ggsave(gp_box, width=10, height=4.5)
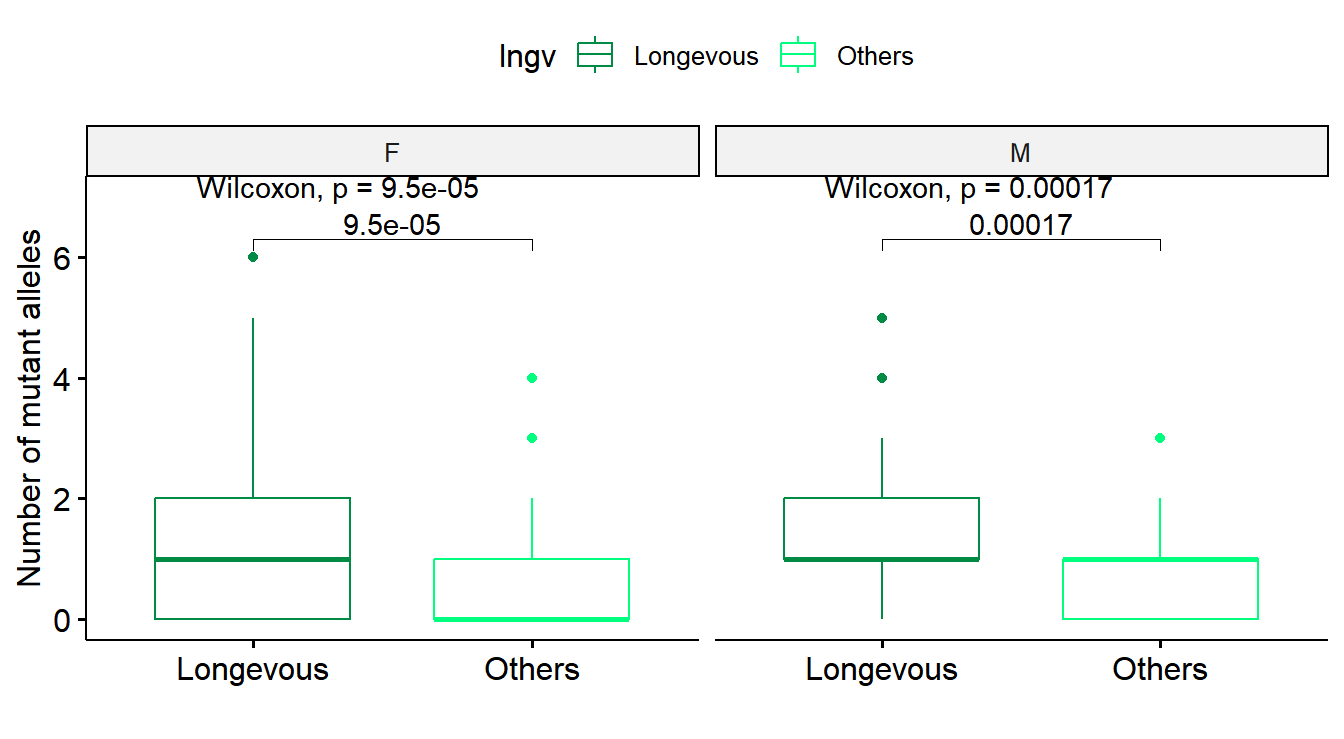
# Figure 2B
# mutant alleles drawn separately by gender
df_tmp1 <- df_all %>% group_by(ID,Sex,Age,agrp,lngv) %>% summarise(nummut=sum(nummut)) %>% ungroup()
gp1 <- df_tmp1 %>% ggboxplot(x="lngv", y="nummut", color="lngv", palette=c("springgreen4","springgreen"))
gp1 <- gp1 + stat_compare_means(comparisons = list(c("Longevous", "Others")), method="wilcox.test") + stat_compare_means(label.y=7) + xlab("") + ylab("Number of mutant alleles")
gp_box1 <- facet(gp1, facet.by="Sex")
# saved into a file 'Boxplot_longevity.pdf'
file.path(outdir,"Boxplot_nummut.pdf") %>% ggsave(gp_box1, width=10, height=4.5)
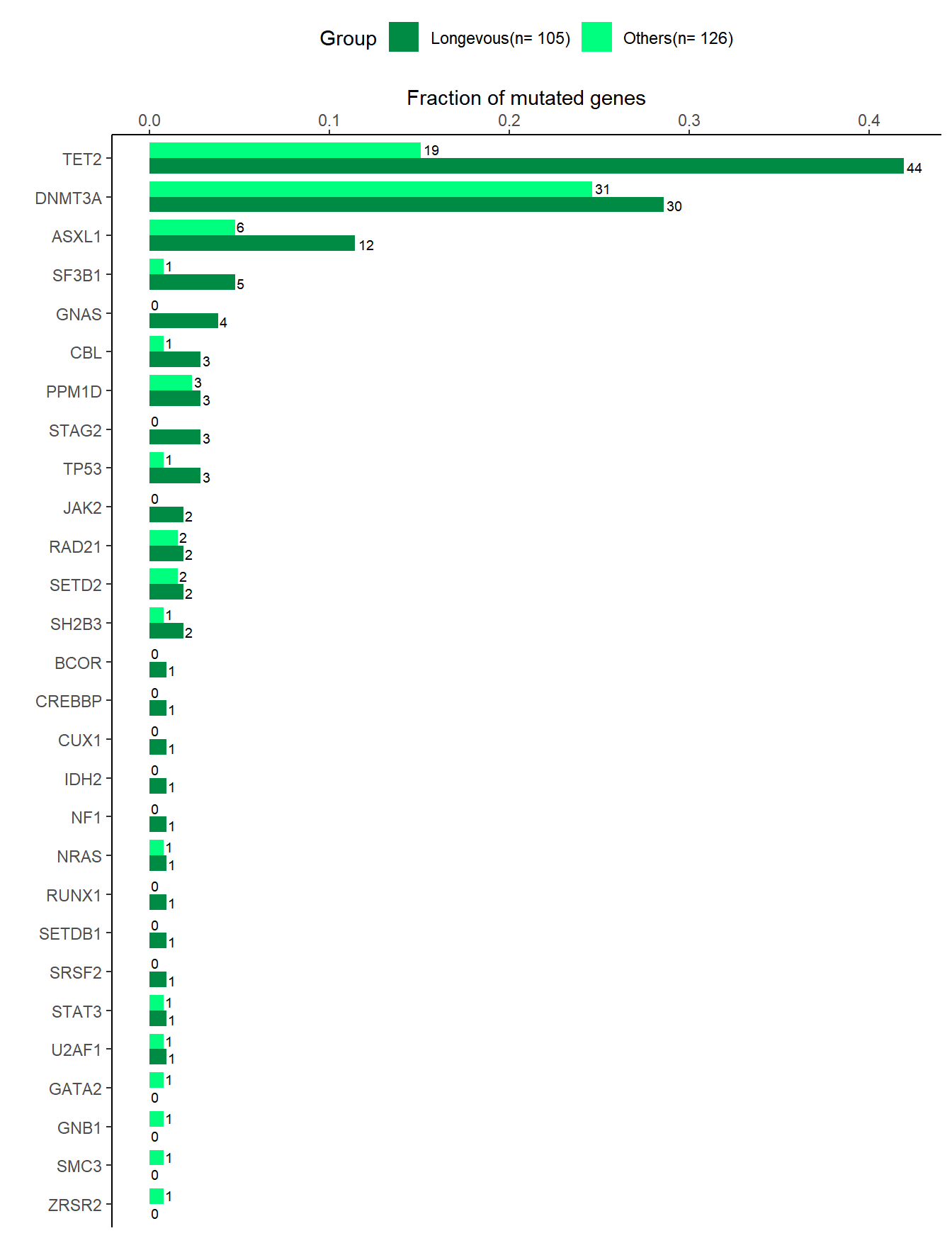
6.2 Number of mutation (barplot)
# Figure 2C (Mutated genes)
# Mutated genes between longevity and others
df_ctrl <- df_input %>% distinct(lngv,gene,ID) %>% count(lngv,gene) %>% rename(total=n)
df_lngv <- df_full %>% filter(!is.na(mutation)) %>% distinct(lngv,gene,ID) %>% count(lngv,gene)
df_ctrl_lngv <- df_ctrl %>% full_join(df_lngv, by=c('lngv','gene')) %>% mutate(n=ifelse(is.na(n),0,n)) %>% mutate(freq=n/total) %>% mutate(Group=str_c(lngv,'(n= ', total,')')) %>% arrange(Group,-freq,gene)
data <- df_ctrl_lngv %>% mutate(gene=factor(gene,levels=unique(gene) %>% rev()))
# plot
p <- data %>% ggplot(aes(x=freq, y=gene, fill=Group))
p <- p + geom_bar(stat="identity", position=position_dodge(), width=0.8) + scale_fill_manual(values=c('springgreen4','springgreen')) + theme_classic() + xlab('Fraction of mutated genes') +ylab("")+ scale_x_continuous(position='top') + theme(legend.position="top")
gp_bar <- p + geom_text(aes(label=n), hjust=-0.2, position=position_dodge(0.9), size=2.5)
# saved into a file 'Barplot_mutated_Genes.pdf'
file.path(outdir,"Barplot_mutated_Genes.pdf") %>% ggsave(gp_bar, width=5, height=7.5)
# Univariate logistic regression (Mutated genes)
df_numgene <- df_all %>% group_by(ID,gene) %>% mutate(numgene=sum(nummut>0)) %>% ungroup()
vec_genes <- df_all %>% distinct(gene) %>% pull(gene)
ls_res <- lapply(vec_genes, function(x){
#x <- 'TET2'
df_tmp <- df_numgene %>% mutate(y=ifelse(Age>=90,1,0)) %>% filter(gene==x)
model <- glm(y ~ Sex + numgene, family="binomial", data=df_tmp)
res1 <- cbind(OR=coef(model), confint(model)) %>% exp()
# ANOVA (Chisq test) (account for gender)
res2 <- anova(model, test="Chisq") %>% as.matrix()
# output
res <- tibble(gene=x, OR=res1[3,1], CIl=res1[3,2], CIh=res1[3,3], pval=res2[3,5])
})
df_res <- do.call(rbind, ls_res)
# at least 2 events per group
df_tmp_gene <- df_ctrl_lngv %>% distinct(lngv,gene,n) %>% pivot_wider(names_from=lngv, values_from=n) %>% filter(`Longevous`>=2, `Others`>=2)
df_res_gene <- df_res %>% semi_join(df_tmp_gene, by='gene')
print(df_res_gene)
## # A tibble: 6 × 5
## gene OR CIl CIh pval
## <chr> <dbl> <dbl> <dbl> <dbl>
## 1 DNMT3A 1.24 0.683 2.25 0.481
## 2 TET2 4.05 2.19 7.75 0.00000594
## 3 ASXL1 3.31 1.19 10.2 0.0209
## 4 PPM1D 1.69 0.298 9.66 0.536
## 5 RAD21 1.58 0.182 13.8 0.656
## 6 SETD2 1.13 0.131 9.70 0.906
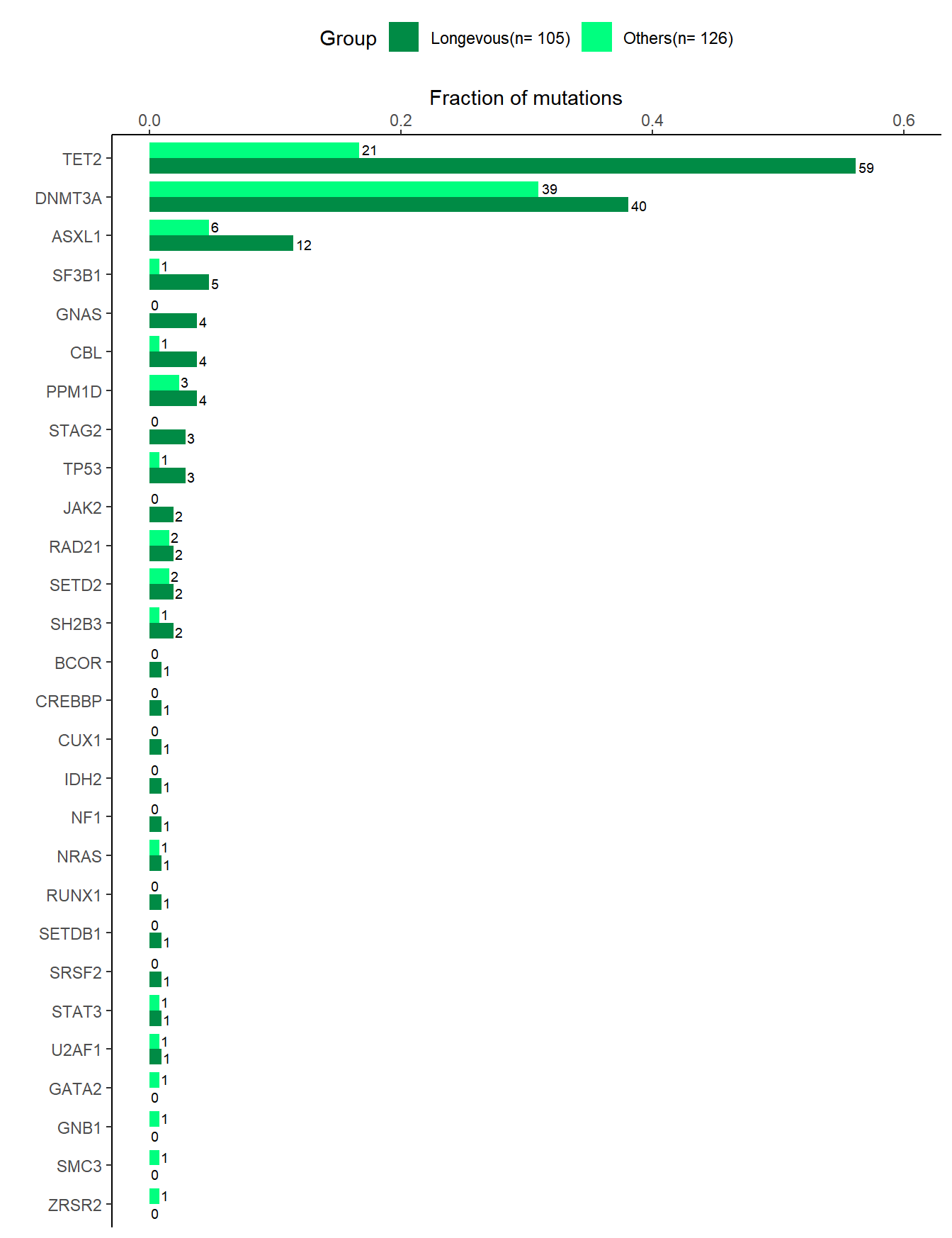
# Figure 2C (Mutant alleles)
# Mutant alleles between longevity and others
df_lngv1 <- df_full %>% group_by(lngv,gene) %>% mutate(vaf=ifelse(is.na(vaf),0,vaf)) %>% summarise(nummut=sum(vaf>0)) #%>% count(lngv,gene) #%>% rename(total=n)
df_ctrl_lngv1 <- df_lngv1 %>% full_join(df_ctrl, by=c('lngv','gene')) %>% mutate(freq=nummut/total) %>% mutate(Group=str_c(lngv,'(n= ', total,')')) %>% arrange(Group,-freq,gene)
data1 <- df_ctrl_lngv1 %>% mutate(gene=factor(gene,levels=levels(data$gene)))
# plot
p1 <- data1 %>% ggplot(aes(x=freq, y=gene, fill=Group))
gp_bar1 <- p1 + geom_bar(stat="identity", position=position_dodge(), width=0.8) + scale_fill_manual(values=c('springgreen4','springgreen')) + theme_classic() + xlab('Fraction of mutations') + ylab("") + scale_x_continuous(position='top',limits = c(0.0,0.6)) + theme(legend.position="top") + geom_text(aes(label=nummut), hjust=-0.2, position=position_dodge(0.9), size=2.5)
# saved into a file 'Barplot_mutant_alleles.pdf'
file.path(outdir,"Barplot_mutant_alleles.pdf") %>% ggsave(gp_bar1, width=5, height=7.5)
# Univariate logistic regression (Mutant alleles).
df_nummut <- df_full %>% group_by(ID,gene) %>% summarize(nummut=sum(vaf>0)) %>% ungroup() %>% mutate(nummut=ifelse(is.na(nummut),0,nummut))
df_nummut1<- df_input %>% inner_join(df_nummut, by=c('ID','gene'))
ls_res1 <- lapply(vec_genes, function(x){
#x <- 'TET2'
df_tmp <- df_nummut1 %>% mutate(y=ifelse(Age>=90,1,0)) %>% filter(gene==x)
model <- glm(y ~ Sex + nummut, family="binomial", data = df_tmp)
res1 <- cbind(OR=coef(model), confint(model)) %>% exp()
res2 <- anova(model, test="Chisq") %>% as.matrix()
res <- tibble(gene=x, OR=res1[3,1], CIl=res1[3,2], CIh=res1[3,3], pval=res2[3,5])
})
df_res1 <- do.call(rbind, ls_res1)
# at least 2 events per group
df_tmp_alleles <- df_ctrl_lngv1 %>% distinct(lngv,gene,nummut) %>% pivot_wider(names_from=lngv, values_from=nummut) %>% filter(`Longevous`>=2, `Others`>=2)
df_res_alleles <- df_res1 %>% semi_join(df_tmp_alleles, by='gene')
print(df_res_alleles)
## # A tibble: 6 × 5
## gene OR CIl CIh pval
## <chr> <dbl> <dbl> <dbl> <dbl>
## 1 DNMT3A 1.21 0.804 1.84 0.357
## 2 TET2 3.17 1.90 5.61 0.00000222
## 3 ASXL1 3.31 1.19 10.2 0.0209
## 4 PPM1D 1.94 0.482 9.27 0.340
## 5 RAD21 1.58 0.182 13.8 0.656
## 6 SETD2 1.13 0.131 9.70 0.906