5 Figure 4

Figure 4: Metabolic information yields an independent prognostic clustering classification for BCP-ALL and T-ALL.
A. The schematic workflow following SNF integration analysis. The BMMC omics data were inputted to generate a fused similarity network, which was subsequently utilized for sample clustering in this study.
B. The discrimination of three BCP-ALL clusters (BC1, BC2 and BC3) generated from SNF method.
C. The association of the three metabolic clusters with clinical OS in 75 BCP-ALL patients.
D. The multivariate Cox analysis reveals that the metabolic cluster bore an independent prognostic value for OS and EFS, respectively. The WBC was stratified by 30×109/L. In the genetic-based risk stratification, BCR::ABL1, ETV6::RUNX1, DUX4::IGH and TCF3::PBX1 were categorized into standard risk subgroup, otherwise were into poor risk subgroup.
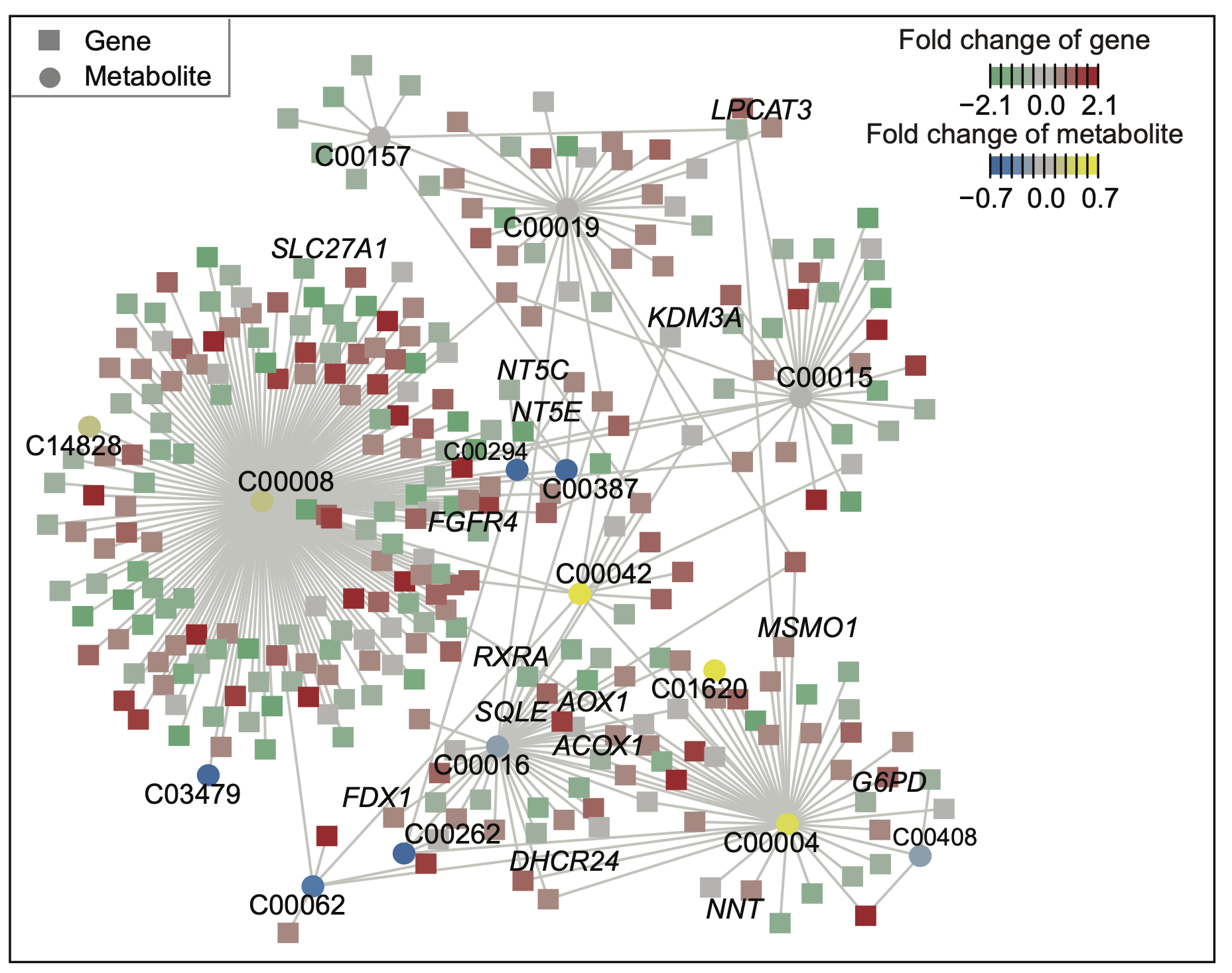
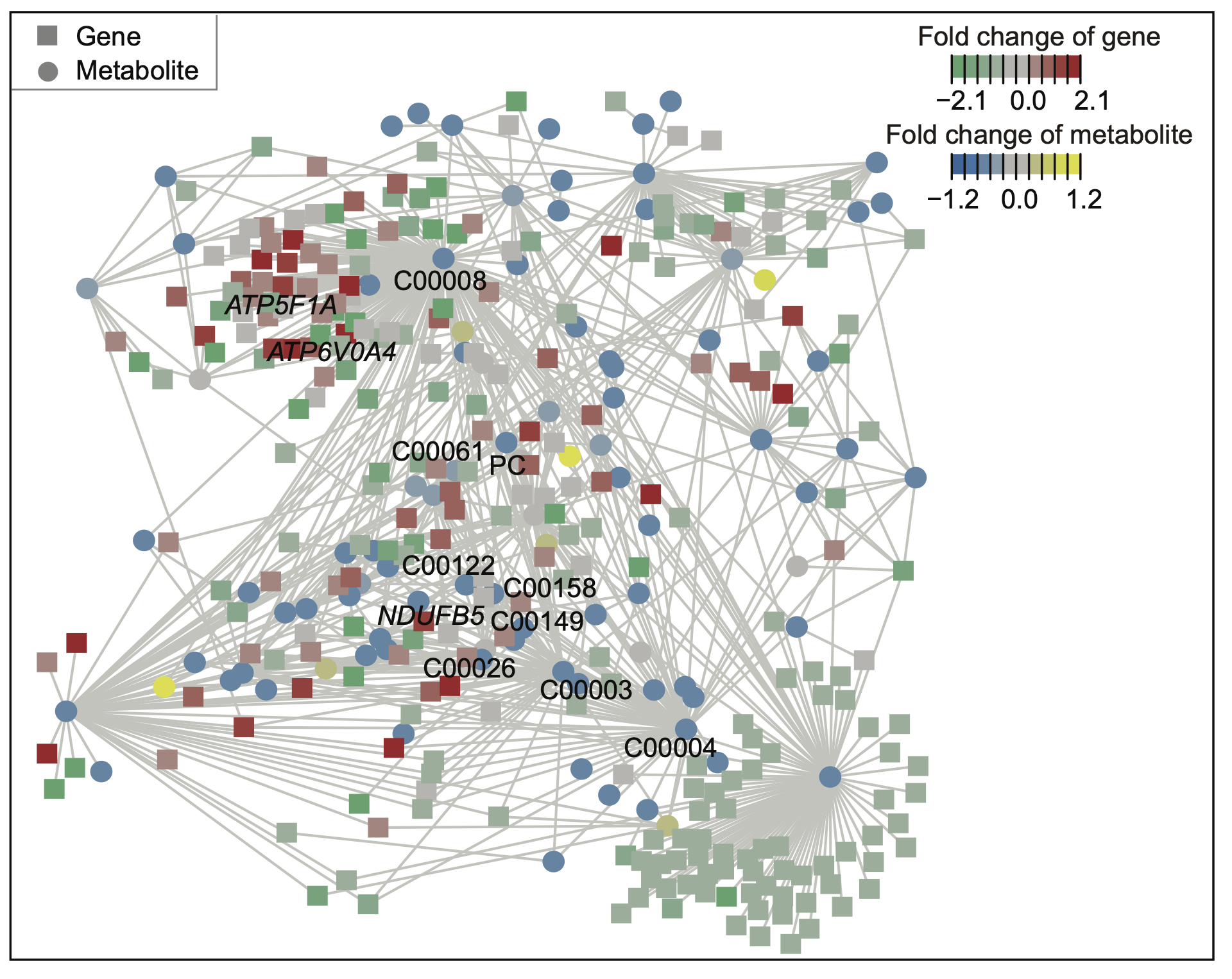
E. A core metabolism-based subnetwork that best explains the difference between BC3 and BC1/2. In this network, each circular node represents a metabolite, labeled with its KEGG ID, and each square node represents a gene. Node colors indicate logFC values: red and green signify different gene expression levels, while yellow and blue signify varying metabolite levels. The highlighted labels correspond to pathways in the cholesterol metabolism and nicotinate and nicotinamide metabolism.
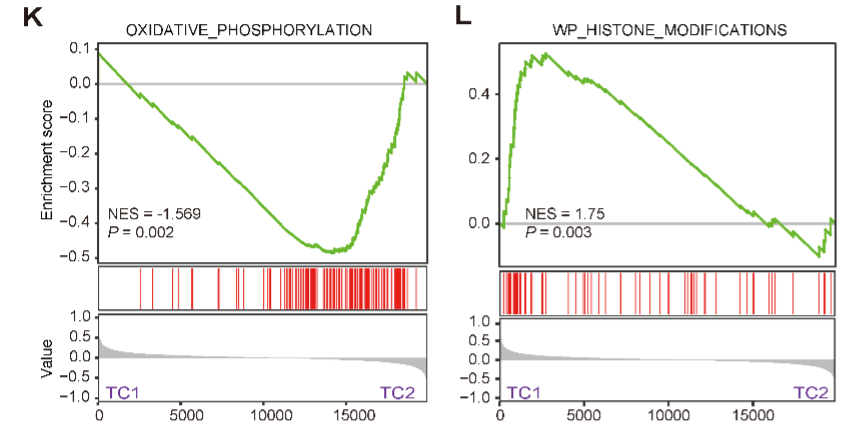
F. The discrimination of two T-ALL clusters (TC1 and TC2) based on SNF method.
G. The association of the two metabolic clusters with clinical OS outcomes in 24 T-ALL patients. The result suggested TC1 was of significantly poorer prognosis than TC2.
H. A core metabolism-based subnetwork that best explains the difference between TC2 and TC1. The highlighted labels correspond to pathways in the TCA and OXPHOS. PC refers to a group of phosphatidylcholines without specific KEGG IDs.
5.1 (A) Schematic Workflow
The schematic workflow following integrated analysis. The BMMC omics data are inputted to generate a fused similarity network, which is subsequently utilized for sample clustering in this study.
library(dplyr)
library(MNet)
library(survival)
library(survminer)
#-------------------------------------------------------------------------------
# Step 1: Cox regression of metabolomics of BCP-ALL
#-------------------------------------------------------------------------------
sample_info <- readxl::read_excel("raw_data/sample_clinical_201_info.xlsx") %>%
as.data.frame() %>%
dplyr::filter(!is.na(METc_BM_D0_ID)) %>%
dplyr::filter(Lineage=="B") %>%
dplyr::mutate(bianhao=gsub("RJ-","A",Pid)) %>%
dplyr::select(bianhao,METc_BM_D0_ID)
dat_cell <- data.table::fread("raw_data/cell_dat_final_reuslt_v0329.txt") %>%
as.data.frame() %>%
tibble::column_to_rownames("label") %>%
dplyr::select(all_of(sample_info$METc_BM_D0_ID)) %>%
log1p()
names(dat_cell) <- sample_info$bianhao
clinical <- readxl::read_excel("raw_data/08 队列病人生存资料-v231026.xlsx") %>%
as.data.frame() %>%
dplyr::select(bianhao,OS_stat,OS) %>%
dplyr::inner_join(sample_info,by=c("bianhao")) %>%
dplyr::arrange(match(bianhao,sample_info$bianhao)) %>%
dplyr::select(-METc_BM_D0_ID) %>%
unique()
dat_t <- t(dat_cell) %>%
data.frame() %>%
tibble::rownames_to_column(var="bianhao") %>%
dplyr::left_join(clinical,by="bianhao") %>%
dplyr::select(OS,OS_stat,everything()) %>%
tibble::column_to_rownames("bianhao") %>%
dplyr::rename("time"="OS") %>%
dplyr::rename("status"="OS_stat")
metabolite_name <- names(dat_t)[3:ncol(dat_t)]
dat_t_raw <- t(dat_cell) %>%
as.data.frame() %>%
tibble::rownames_to_column(var="bianhao") %>%
dplyr::left_join(clinical,by="bianhao") %>%
dplyr::select(OS,OS_stat,everything()) %>%
tibble::column_to_rownames("bianhao") %>%
dplyr::rename("time"="OS") %>%
dplyr::rename("status"="OS_stat")
metabolite_name_raw <- names(dat_t_raw)[3:ncol(dat_t_raw)]
dd <- data.frame(name=metabolite_name,name_raw=metabolite_name_raw)
univ_formulas <- lapply(metabolite_name,function(x) stats::as.formula(paste('survival::Surv(time, status)~', x)))
univ_models <- lapply( univ_formulas, function(x){survival::coxph(x, data = dat_t)})
univ_results <- lapply(univ_models,
function(x){
x <- summary(x)
p.value<-signif(x$wald["pvalue"], digits=2)
wald.test<-signif(x$wald["test"], digits=2)
beta<-signif(x$coef[1], digits=2);#coeficient beta
HR <-signif(x$coef[2], digits=2);#exp(beta)
HR.confint.lower <- signif(x$conf.int[,"lower .95"], 2)
HR.confint.upper <- signif(x$conf.int[,"upper .95"],2)
HR <- paste0(HR, " (",
HR.confint.lower, "-", HR.confint.upper, ")")
name <- rownames(x$coefficients)
res<-c(name,beta, HR, wald.test, p.value)
names(res)<-c("name","beta", "HR (95% CI for HR)", "wald.test","p.value")
return(res)
})
res <- t(as.data.frame(univ_results, check.names = FALSE))
result <- as.data.frame(res)
metabolite_all <- result %>%
dplyr::left_join(dd,by=c("name"))
write.table(metabolite_all,"result/Figure4/4A_1.cox_metabolite_all_B.txt",quote=F,row.names=F,sep="\t")
#-------------------------------------------------------------------------------
# Step 2: Cox regression of transcriptomics of BCP-ALL
#-------------------------------------------------------------------------------
gene_metabolite1 <- gene_metabolite %>% dplyr::filter(dest_type=="gene")
gene2 <- PathwayExtendData %>% filter(type=="gene")
gene_filter <- unique(c(gene_metabolite1$gene,gene2$name))
length(gene_filter)
sample_rna <- readxl::read_excel("raw_data/sample_clinical_201_info.xlsx") %>%
as.data.frame() %>%
dplyr::filter(Lineage=="B") %>%
dplyr::filter(!is.na(RNA_ID)) %>%
dplyr::mutate(bianhao=gsub("RJ-","A",Pid)) %>%
dplyr::select(bianhao,RNA_id_raw)
dat_gene <- readRDS("raw_data/20231113_RNA_ALL_VST_coding.rds") %>%
as.data.frame() %>%
dplyr::select(sample_rna$RNA_id_raw) %>%
tibble::rownames_to_column(var="label") %>%
dplyr::filter(label %in% gene_filter) %>%
tibble::column_to_rownames("label")
names(dat_gene) <- sample_rna$bianhao
dat_gene_0 <- rowSums(dat_gene==0)
dat_gene <- dat_gene %>%
dplyr::mutate(num=dat_gene_0) %>%
dplyr::filter(num < 0.5*ncol(dat_gene)) %>%
dplyr::select(-num)
clinical <- readxl::read_excel("raw_data/08 队列病人生存资料-v231026.xlsx") %>%
as.data.frame() %>%
dplyr::select(bianhao_new,OS_stat,OS) %>%
dplyr::mutate(bianhao=paste0("A",bianhao_new)) %>%
dplyr::select(-bianhao_new) %>%
unique()
dat_t <- t(dat_gene) %>%
data.frame() %>%
tibble::rownames_to_column(var="bianhao") %>%
dplyr::left_join(clinical,by="bianhao") %>%
dplyr::select(OS,OS_stat,everything()) %>%
tibble::column_to_rownames("bianhao") %>%
dplyr::rename("time"="OS") %>%
dplyr::rename("status"="OS_stat")
gene_name <- names(dat_t)[3:ncol(dat_t)]
dat_t_raw <- t(dat_gene) %>%
as.data.frame() %>%
tibble::rownames_to_column(var="bianhao") %>%
dplyr::left_join(clinical,by="bianhao") %>%
dplyr::select(OS,OS_stat,everything()) %>%
tibble::column_to_rownames("bianhao") %>%
dplyr::rename("time"="OS") %>%
dplyr::rename("status"="OS_stat")
gene_name_raw <- names(dat_t_raw)[3:ncol(dat_t_raw)]
dd <- data.frame(name=gene_name,name_raw=gene_name_raw)
univ_formulas <- lapply(gene_name,function(x) stats::as.formula(paste('survival::Surv(time, status)~', x)))
univ_models <- lapply(univ_formulas, function(x){survival::coxph(x, data = dat_t)})
univ_results <- lapply(univ_models,
function(x){
x <- summary(x)
p.value<-signif(x$wald["pvalue"], digits=2)
wald.test<-signif(x$wald["test"], digits=2)
beta<-signif(x$coef[1], digits=2);#coeficient beta
HR <-signif(x$coef[2], digits=2);#exp(beta)
HR.confint.lower <- signif(x$conf.int[,"lower .95"], 2)
HR.confint.upper <- signif(x$conf.int[,"upper .95"],2)
HR <- paste0(HR, " (",
HR.confint.lower, "-", HR.confint.upper, ")")
name <- rownames(x$coefficients)
res<-c(name,beta, HR, wald.test, p.value)
names(res)<-c("name","beta", "HR (95% CI for HR)", "wald.test","p.value")
return(res)
})
res <- t(as.data.frame(univ_results, check.names = FALSE))
result <- as.data.frame(res)
gene_all <- result %>%
dplyr::left_join(dd,by="name")
write.table(gene_all,"result/Figure4/4A_1.cox_gene_all_B.txt",quote=F,row.names=F,sep="\t")
#------------------------------------------------------------------------------
# Step 3: Cox regression of metabolomics of T-ALL
#------------------------------------------------------------------------------
sample_info <- readxl::read_excel("raw_data/sample_clinical_201_info.xlsx") %>%
as.data.frame() %>%
dplyr::filter(!is.na(METc_BM_D0_ID)) %>%
dplyr::filter(Lineage=="T") %>%
dplyr::mutate(bianhao=gsub("RJ-","A",Pid)) %>%
dplyr::select(bianhao,METc_BM_D0_ID)
dat_cell <- data.table::fread("raw_data/cell_dat_final_reuslt_v0329.txt") %>%
as.data.frame() %>%
tibble::column_to_rownames("label") %>%
dplyr::select(all_of(sample_info$METc_BM_D0_ID)) %>%
log1p()
names(dat_cell) <- sample_info$bianhao
clinical <- readxl::read_excel("raw_data/08 队列病人生存资料-v231026.xlsx") %>%
as.data.frame() %>%
dplyr::select(bianhao,OS_stat,OS) %>%
dplyr::inner_join(sample_info,by=c("bianhao")) %>%
dplyr::arrange(match(bianhao,sample_info$bianhao)) %>%
dplyr::select(-METc_BM_D0_ID) %>%
unique()
dat_t <- t(dat_cell) %>%
data.frame() %>%
tibble::rownames_to_column(var="bianhao") %>%
dplyr::left_join(clinical,by="bianhao") %>%
dplyr::select(OS,OS_stat,everything()) %>%
tibble::column_to_rownames("bianhao") %>%
dplyr::rename("time"="OS") %>%
dplyr::rename("status"="OS_stat")
metabolite_name <- names(dat_t)[3:ncol(dat_t)]
dat_t_raw <- t(dat_cell) %>%
as.data.frame() %>%
tibble::rownames_to_column(var="bianhao") %>%
dplyr::left_join(clinical,by="bianhao") %>%
dplyr::select(OS,OS_stat,everything()) %>%
tibble::column_to_rownames("bianhao") %>%
dplyr::rename("time"="OS") %>%
dplyr::rename("status"="OS_stat")
metabolite_name_raw <- names(dat_t_raw)[3:ncol(dat_t_raw)]
dd <- data.frame(name=metabolite_name,name_raw=metabolite_name_raw)
univ_formulas <- lapply(metabolite_name,function(x) stats::as.formula(paste('survival::Surv(time, status)~', x)))
univ_models <- lapply( univ_formulas, function(x){survival::coxph(x, data = dat_t)})
univ_results <- lapply(univ_models,
function(x){
x <- summary(x)
p.value<-signif(x$wald["pvalue"], digits=2)
wald.test<-signif(x$wald["test"], digits=2)
beta<-signif(x$coef[1], digits=2);
HR <-signif(x$coef[2], digits=2);
HR.confint.lower <- signif(x$conf.int[,"lower .95"], 2)
HR.confint.upper <- signif(x$conf.int[,"upper .95"],2)
HR <- paste0(HR, " (",
HR.confint.lower, "-", HR.confint.upper, ")")
name <- rownames(x$coefficients)
res<-c(name,beta, HR, wald.test, p.value)
names(res)<-c("name","beta", "HR (95% CI for HR)", "wald.test","p.value")
return(res)
})
res <- t(as.data.frame(univ_results, check.names = FALSE))
result <- as.data.frame(res)
metabolite_all <- result %>%
dplyr::left_join(dd,by=c("name"))
write.table(metabolite_all,"result/Figure4/4A_1.cox_metabolite_all_T.txt",quote=F,row.names=F,sep="\t")
#-------------------------------------------------------------------------------
# Step 4: Cox regression of transcriptomics of T-ALL
#-------------------------------------------------------------------------------
gene_metabolite1 <- gene_metabolite %>% dplyr::filter(dest_type=="gene")
gene2 <- PathwayExtendData %>% filter(type=="gene")
gene_filter <- unique(c(gene_metabolite1$gene,gene2$name))
length(gene_filter)
sample_rna <- readxl::read_excel("raw_data/sample_clinical_201_info.xlsx") %>%
as.data.frame() %>%
dplyr::filter(Lineage=="T") %>%
dplyr::filter(!is.na(RNA_ID)) %>%
dplyr::mutate(bianhao=gsub("RJ-","A",Pid)) %>%
dplyr::select(bianhao,RNA_id_raw)
dat_gene <- readRDS("raw_data/20231113_RNA_ALL_VST_coding.rds") %>%
as.data.frame() %>%
dplyr::select(sample_rna$RNA_id_raw) %>%
tibble::rownames_to_column(var="label") %>%
dplyr::filter(label %in% gene_filter) %>%
tibble::column_to_rownames("label")
names(dat_gene) <- sample_rna$bianhao
dat_gene_0 <- rowSums(dat_gene==0)
dat_gene <- dat_gene %>%
dplyr::mutate(num=dat_gene_0) %>%
dplyr::filter(num < 0.5*ncol(dat_gene)) %>%
dplyr::select(-num)
clinical <- readxl::read_excel("raw_data/08 队列病人生存资料-v231026.xlsx") %>%
as.data.frame() %>%
dplyr::select(bianhao_new,OS_stat,OS) %>%
dplyr::mutate(bianhao=paste0("A",bianhao_new)) %>%
dplyr::select(-bianhao_new) %>%
unique()
dat_t <- t(dat_gene) %>%
data.frame() %>%
tibble::rownames_to_column(var="bianhao") %>%
dplyr::left_join(clinical,by="bianhao") %>%
dplyr::select(OS,OS_stat,everything()) %>%
tibble::column_to_rownames("bianhao") %>%
dplyr::rename("time"="OS") %>%
dplyr::rename("status"="OS_stat")
gene_name <- names(dat_t)[3:ncol(dat_t)]
dat_t_raw <- t(dat_gene) %>%
as.data.frame() %>%
tibble::rownames_to_column(var="bianhao") %>%
dplyr::left_join(clinical,by="bianhao") %>%
dplyr::select(OS,OS_stat,everything()) %>%
tibble::column_to_rownames("bianhao") %>%
dplyr::rename("time"="OS") %>%
dplyr::rename("status"="OS_stat")
gene_name_raw <- names(dat_t_raw)[3:ncol(dat_t_raw)]
dd <- data.frame(name=gene_name,name_raw=gene_name_raw)
univ_formulas <- lapply(gene_name,function(x) stats::as.formula(paste('survival::Surv(time, status)~', x)))
univ_models <- lapply( univ_formulas, function(x){survival::coxph(x, data = dat_t)})
univ_results <- lapply(univ_models,
function(x){
x <- summary(x)
p.value<-signif(x$wald["pvalue"], digits=2)
wald.test<-signif(x$wald["test"], digits=2)
beta<-signif(x$coef[1], digits=2);#coeficient beta
HR <-signif(x$coef[2], digits=2);#exp(beta)
HR.confint.lower <- signif(x$conf.int[,"lower .95"], 2)
HR.confint.upper <- signif(x$conf.int[,"upper .95"],2)
HR <- paste0(HR, " (",
HR.confint.lower, "-", HR.confint.upper, ")")
name <- rownames(x$coefficients)
res<-c(name,beta, HR, wald.test, p.value)
names(res)<-c("name","beta", "HR (95% CI for HR)", "wald.test",
"p.value")
return(res)
})
res <- t(as.data.frame(univ_results, check.names = FALSE))
result <- as.data.frame(res)
gene_all <- result %>%
dplyr::left_join(dd,by="name")
write.table(gene_all,"result/Figure4/4A_1.cox_gene_all_T.txt",quote=F,row.names=F,sep="\t")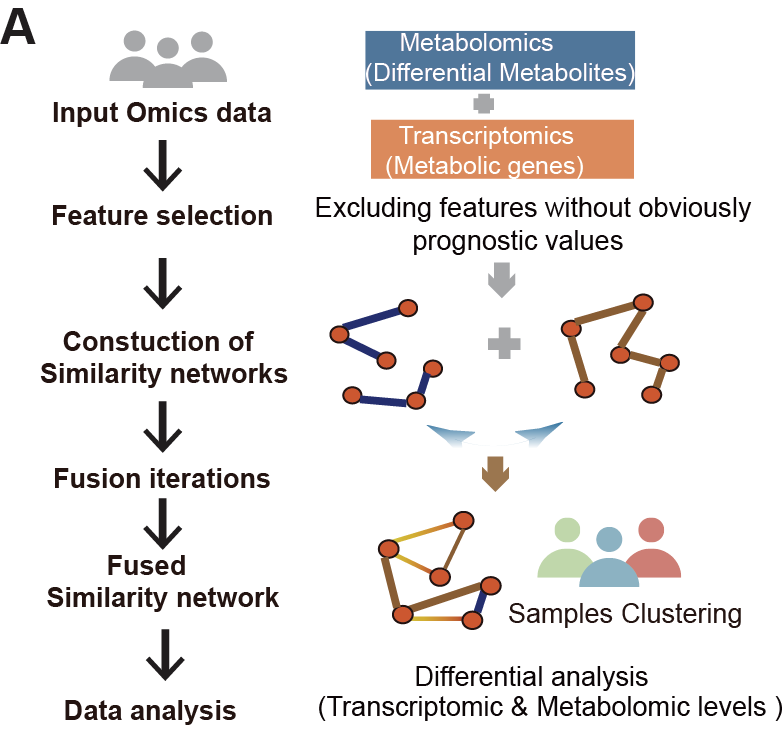
5.2 (B) Discrimination of BCP-ALL Clusters
The discrimination of three clusters (BC1, BC2 and BC3) generated from SNF method.
library(dplyr)
library(MNet)
library(survival)
library(survminer)
#-------------------------------------------------------------------------------
# Step 1: Load data and set parameters
#-------------------------------------------------------------------------------
dat_filter <- data.table::fread("result/Figure2/1.BMMCvsHC.txt") %>%
as.data.frame() %>%
dplyr::arrange(P.Value) %>%
dplyr::filter(P.Value < 0.5)
dat_filter$P.Value[1:5]
dat_cox <- data.table::fread("result/Figure4/4A_1.cox_metabolite_all_B.txt") %>%
as.data.frame() %>%
dplyr::arrange(p.value) %>%
head(n=120)
dat_cox$p.value[1:5]
dat_filter <- dat_filter %>%
dplyr::filter(name %in% dat_cox$name_raw)
sample_info <- readxl::read_excel("raw_data/sample_clinical_201_info.xlsx") %>%
as.data.frame() %>%
dplyr::filter(!is.na(METc_BM_D0_ID)) %>%
dplyr::filter(!is.na(RNA_ID)) %>%
dplyr::filter(Lineage=="B")
dat_cell <- data.table::fread("raw_data/cell_dat_final_reuslt_v0329.txt") %>%
as.data.frame() %>%
dplyr::filter(label %in% dat_filter$name) %>%
tibble::column_to_rownames("label") %>%
dplyr::select(all_of(sample_info$METc_BM_D0_ID)) %>%
log1p()
names(dat_cell) <- sample_info$Pid
cox_gene <- data.table::fread("result/Figure4/4A_1.cox_gene_all_B.txt") %>%
as.data.frame() %>%
dplyr::arrange(p.value) %>%
head(n=600)
dat_gene <- readRDS("raw_data/20231113_RNA_ALL_VST_coding.rds") %>%
as.data.frame() %>%
dplyr::select(sample_info$RNA_id_raw) %>%
tibble::rownames_to_column(var="label") %>%
dplyr::filter(label %in% cox_gene$name_raw) %>%
tibble::column_to_rownames("label")
names(dat_gene) <- sample_info$Pid
#-------------------------------------------------------------------------------
# Step 2: SNF of discrimination BCP-ALL clusters
#-------------------------------------------------------------------------------
library(SNFtool)
metabolite_norm = standardNormalization(t(dat_cell))
metgene_norm = standardNormalization(t(dat_gene))
dim(metabolite_norm)
dim(metgene_norm)
write.table(metabolite_norm,"result/Figure4/4B_metabolite_norm_B.txt",quote=F,sep="\t")
write.table(metgene_norm,"result/Figure4/4B_metgene_norm_B.txt",quote=F,sep="\t")
Dist1 = (SNFtool::dist2(as.matrix(metabolite_norm),as.matrix(metabolite_norm)))^(1/2)
Dist2 = (SNFtool::dist2(as.matrix(metgene_norm),as.matrix(metgene_norm)))^(1/2)
W1 = affinityMatrix(Dist1, 10, 0.5)
W2 = affinityMatrix(Dist2, 10, 0.5)
W = SNF(list(W1,W2), 10, 10)
Clusters = spectralClustering(W,K=3)
M_label=cbind(Clusters,sample_info) %>%
dplyr::mutate(Clusters=ifelse(Clusters==1,"BC3",
ifelse(Clusters==2,"BC2",
ifelse(Clusters==3,"BC1",Clusters))))
write.table(M_label,paste0("result/Figure4/4B_cluster_B.txt"),quote=F,row.names = F,sep="\t")
library(ComplexHeatmap)
l <- M_label %>%
dplyr::arrange(Clusters)
W_2 <- W %>%
as.data.frame() %>%
dplyr::select(l$Pid) %>%
tibble::rownames_to_column(var="sample") %>%
dplyr::arrange(match(sample,l$Pid)) %>%
tibble::column_to_rownames("sample")
W_2[W_2==0.5] <- 0
col_fun = circlize::colorRamp2(c( 0, 0.03), c( "white","#F48326"))
p_metabolite <- Heatmap(W_2,height=unit(10,"cm"),name="Sample Data",
cluster_rows = F,cluster_columns = F,
col = col_fun,
show_column_names = F,show_row_names = F,
row_names_gp = gpar(fontsize = 3),
column_names_gp = gpar(fontsize = 3))
pdf("result/Figure4/4B_SNF_discrimination_complexheatmap_B.pdf",width=5,height = 5)
p_metabolite
dev.off()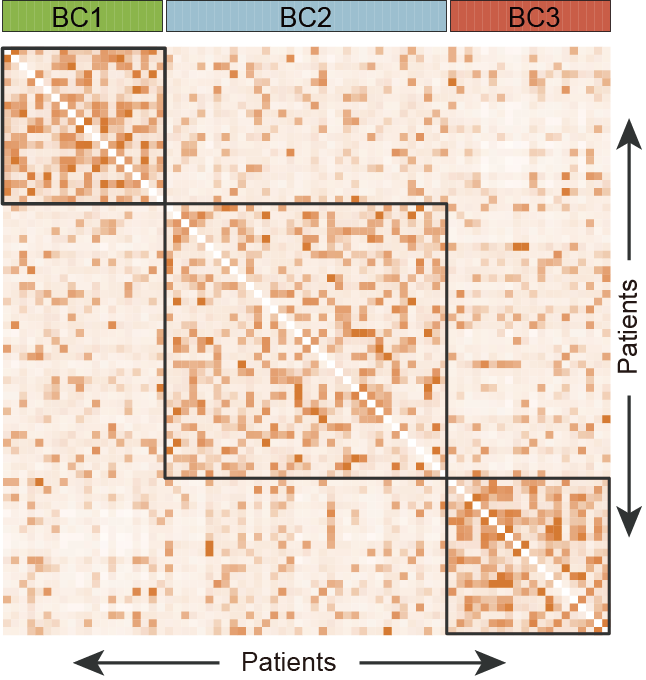
5.3 (C) OS in 75 BCP-ALL patients
The association of the three metabolic clusters with clinical OS in 75 BCP-ALL patients.
library(dplyr)
library(MNet)
library(survival)
library(survminer)
#-------------------------------------------------------------------------------
# Step 1: Load data
#-------------------------------------------------------------------------------
##### new survival #####
survival_new <- readxl::read_excel("raw_data/Meta_Files.xlsx",skip=1) %>%
as.data.frame() %>%
dplyr::select(PID,OS_stat,`OS (days)`) %>%
dplyr::rename("OS"="OS (days)")
clinical_survival <- data.table::fread(paste0("result/Figure4/4B_cluster_B.txt")) %>%
as.data.frame() %>%
dplyr::select(-OS_stat,-OS,-EFS_stat,-EFS,-RFS_stat,-RFS) %>%
left_join(survival_new,by=c("Pid"="PID"))
#-------------------------------------------------------------------------------
# Step 2: Overall survival analysis
#-------------------------------------------------------------------------------
surv_object <- Surv(time = clinical_survival$OS/30, event = clinical_survival$OS_stat)
fit_cluster <- survfit(surv_object ~ Clusters, data = clinical_survival)
p1 <- survminer::ggsurvplot(fit_cluster, data = clinical_survival,
title="OS",
palette=c("#9BD53F","#A6CEE3","#EA644C"),
tables.theme = theme_void(),risk.table = TRUE,
risk.table.fontsize=6,
break.time.by = 12,
risk.table.y.text = FALSE,
tables.height = 0.15,risk.table.col = "black",
#pval = TRUE,pval.method = F,
#pval.coord=c(0.5,0.1),pval.size=6,
font.tickslab=c(14,"plain","black"),
xlab="Time (Months) ",ylab="OS Survival")
pdf(paste0("result/Figure4/4C_OS_B.pdf"),onefile=FALSE)
p1
dev.off()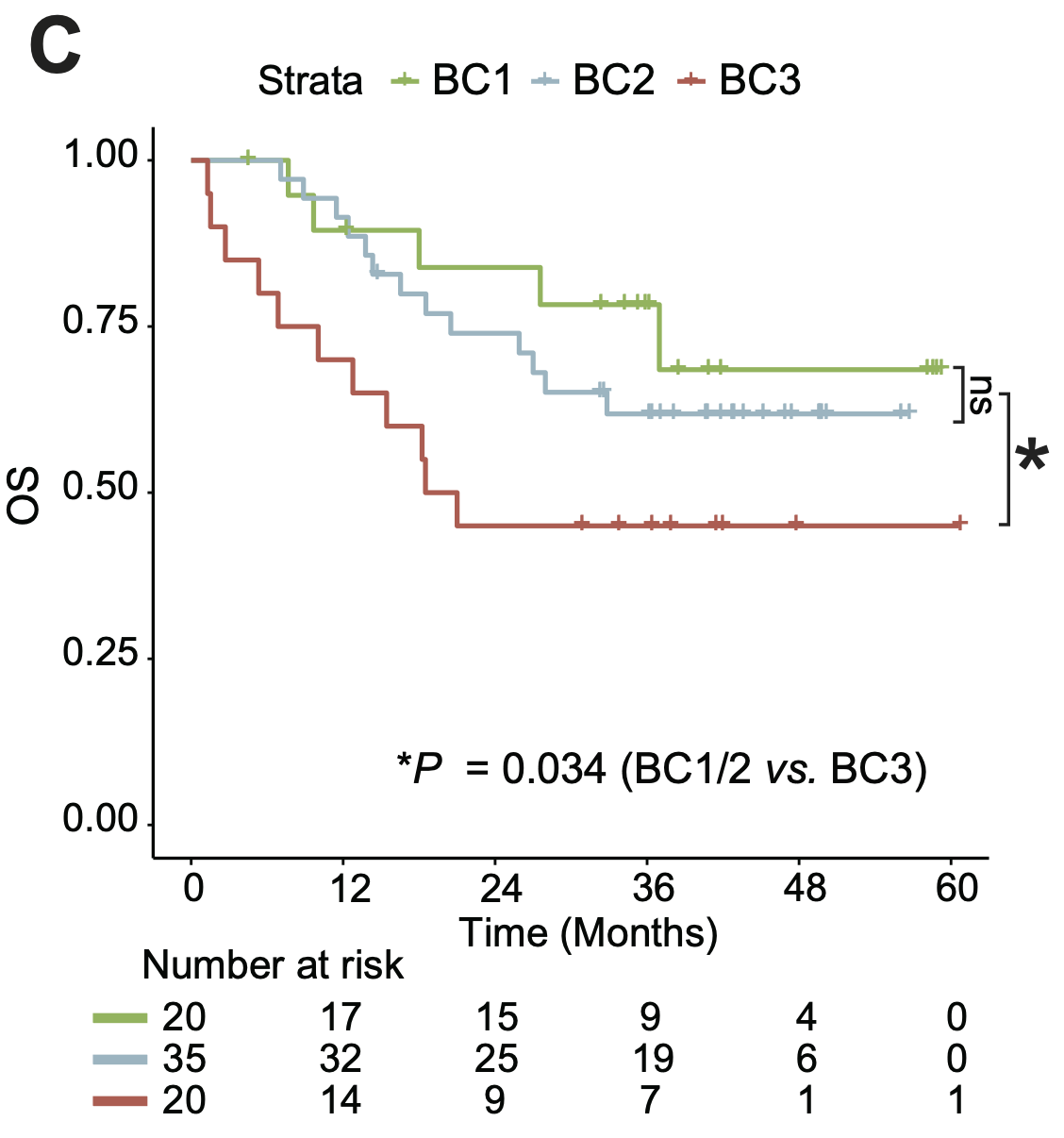
5.4 (D) Multivariate Cox Analysis
The multivariate Cox analysis reveals that the metabolic cluster bore an independent prognostic value for OS and EFS, respectively. The WBC was stratified by 30×109/L. In the genetic-based risk stratification, BCR::ABL1, ETV6::RUNX1, DUX4::IGH and TCF3::PBX1 were categorized into standard risk subgroup, otherwise were into poor risk subgroup.
library(dplyr)
library(openxlsx)
library(survival)
library(survminer)
#-------------------------------------------------------------------------------
# Step 1: Load data and set parameters
#-------------------------------------------------------------------------------
tt <- readxl::read_excel("raw_data/Sample_RNA_190_fusion_mutation_v1130.xlsx") %>%
as.data.frame() %>%
dplyr::select(mutation,fusion,RNA_id,Prediction_Check)
M_label <- data.table::fread("result/Figure4/4B_cluster_B.txt") %>%
as.data.frame() %>%
dplyr::mutate(Clusters=ifelse(Clusters %in% c("BC1","BC2"),"BC1+BC2",Clusters)) %>%
dplyr::mutate(Clusters=as.factor(Clusters)) %>%
dplyr::select(Clusters,Pid)
##### new survival #####
clinical_survival <- readxl::read_excel("raw_data/Meta_Files.xlsx",skip=1) %>%
as.data.frame() %>%
dplyr::rename("OS"="OS (days)","EFS"="EFS (days)","RFS"="RFS (days)","ifHSCT"="HSCT_stat",
"WBC"="WBC (10⁹/L)","Age"="Age (yrs)","Pid"="PID","RNA_id_raw"="RNA_ID_raw") %>%
dplyr::select(Pid,RNA_id_raw,Age,Sex,WBC,ifHSCT,OS,OS_stat,EFS,EFS_stat,RFS,RFS_stat,D28_status) %>%
dplyr::mutate(WBC_type= ifelse(WBC>30, "high",
ifelse(WBC<=30,"low",NA))) %>%
dplyr::mutate(ifHSCT=as.factor(ifHSCT)) %>%
dplyr::inner_join(M_label,by="Pid") %>%
dplyr::left_join(tt,by=c("RNA_id_raw"="RNA_id")) %>%
dplyr::mutate(NCCN=ifelse(fusion %in% c("ETV6-RUNX1","DUX4-IGH","TCF3-PBX1"),"Standard risk",
ifelse(fusion %in% c("KMT2A-AFF1","EP300-ZNF384","SMARCA2-ZNF384"),"Poor risk",
ifelse(Prediction_Check=="Ph-like","Poor risk",
ifelse(Prediction_Check %in% c("BCL2/MYC","PAX5alt","High hyperdoploid", "Low hypodiploid","IDH","TWIST2-high"),"Poor risk","Standard risk"))))) %>%
dplyr::mutate(NCCN=factor(NCCN,levels=c("Standard risk","Poor risk"))) %>%
dplyr::mutate(Sex=factor(Sex,levels=c("M","F"))) %>%
dplyr::mutate(Age=ifelse(Age>40,"old","young")) %>%
dplyr::mutate(Age=factor(Age,levels=c("young","old")))
var.names <- c("Clusters","WBC_type","Age","NCCN","D28_status","ifHSCT")
#-------------------------------------------------------------------------------
# Step 2: Event-free survival (EFS)
#-------------------------------------------------------------------------------
f_efs <- as.formula(paste('Surv(EFS, EFS_stat)~',paste(var.names,collapse = "+")))
fit.final <- coxph(f_efs,data=clinical_survival)
p_efs <- ggforest(fit.final,main="EFS Hzard Ratio",
cpositions=c(0.02,0.22,0.4),
fontsize=0.8,refLabel="reference",
noDigits=2,data=clinical_survival)
#-------------------------------------------------------------------------------
# Step 3: Overall survival(OS)
#-------------------------------------------------------------------------------
f_os <- as.formula(paste('Surv(OS, OS_stat)~',paste(var.names,collapse = "+")))
fit.os <- coxph(f_os,data=clinical_survival)
p_os <- ggforest(fit.os,main="OS Hzard Ratio",
cpositions=c(0.02,0.22,0.4),
fontsize=0.8,refLabel="reference",
noDigits=2,data=clinical_survival)
ggsave(p_efs,filename = "result/Figure4/4D_HR_EFS_B_NCCN_1plus2vs3.pdf",width=9,height=4.5)
ggsave(p_os,filename = "result/Figure4/4D_HR_OS_B_NCCN_1plus2vs3.pdf",width=9,height=4.5)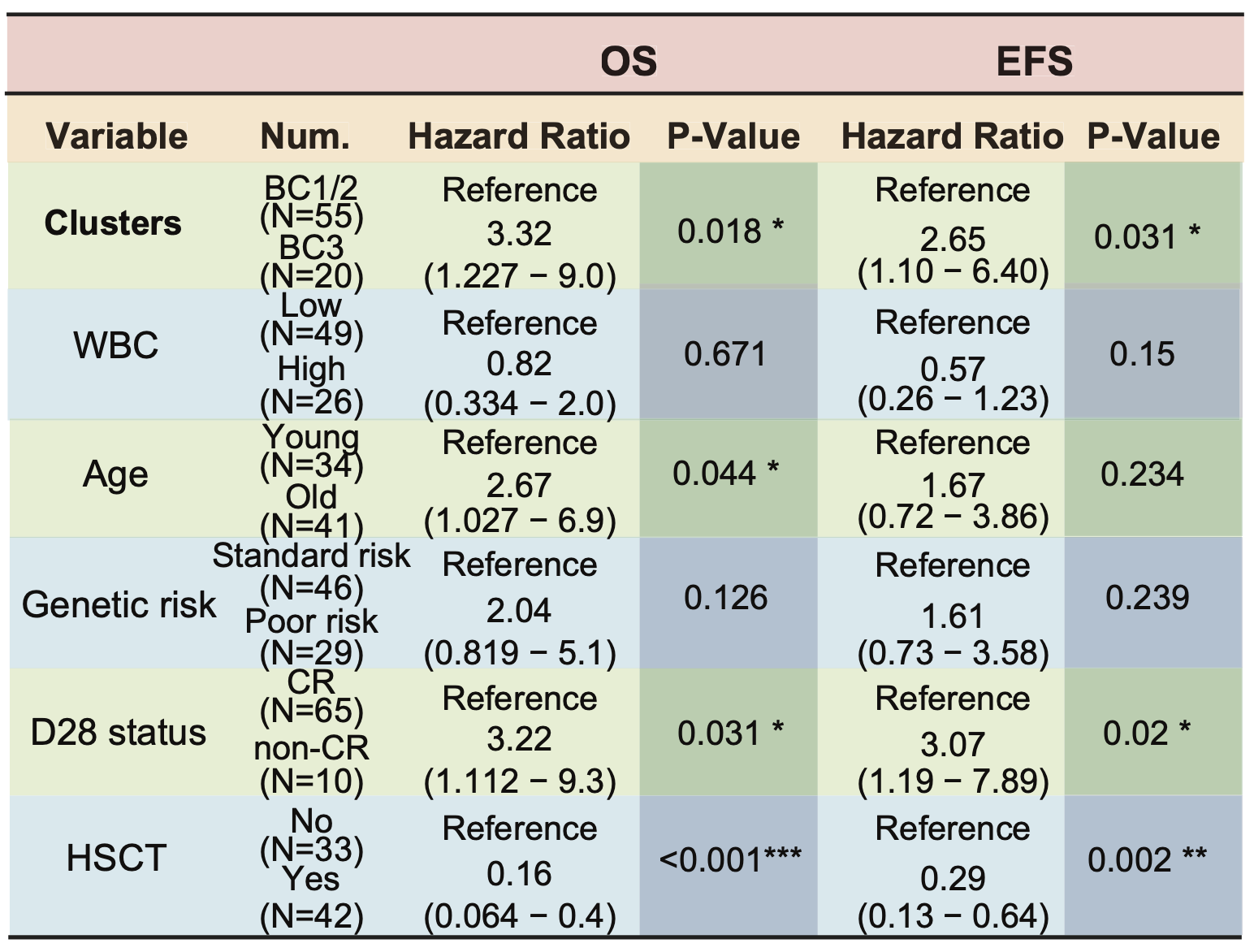
5.5 (E) Subnetwork between BC3 and BC1/2
A core metabolism-based subnetwork that best explains the difference between BC3 and BC1/2. In this network, each circular node represents a metabolite, labeled with its KEGG ID, and each square node represents a gene. Node colors indicate logFC values: red and green signify different gene expression levels, while yellow and blue signify varying metabolite levels. The highlighted labels correspond to pathways in the cholesterol metabolism and nicotinate and nicotinamide metabolism.
library(dplyr)
library(ggplot2)
library(igraph)
library(MNet)
library(graphics)
library(cowplot)
rm(list = ls())
#-------------------------------------------------------------------------------
# Step 1: Read data and set parameters
#-------------------------------------------------------------------------------
gene1 <- readxl::read_excel("raw_data/Chol_Transport_Deg_Ester.xlsx") %>%
as.data.frame()
name_gene <- data.table::fread("result/Figure4/4A_1.cox_gene_all_B.txt") %>%
as.data.frame()
diff_gene <- data.table::fread("result/Figure4/S5H_gene_B.txt") %>%
as.data.frame() %>%
filter(name %in% name_gene$name_raw)
kid <- openxlsx::read.xlsx("raw_data/cell_metabolite_info_all_v1109.xlsx") %>%
as.data.frame() %>%
dplyr::select(refmet_name,KEGG) %>%
mutate(KEGG=ifelse(KEGG=="None",refmet_name,KEGG))
diff_meta <- data.table::fread("result/Figure4/S5H_metabolite_B.txt") %>%
as.data.frame() %>%
inner_join(kid,by=c("name"="refmet_name")) %>%
dplyr::select(-name) %>%
dplyr::rename("name"="KEGG") %>%
arrange(P.Value) %>%
distinct(name,.keep_all = T)
names(diff_meta)[4] <- "p_value"
names(diff_gene)[4] <- "p_value"
dd <- data.table::fread("raw_data/subnetwork_important_edge-300_B.txt") %>%
as.data.frame()
set.seed(1)
g <- graph_from_data_frame(dd)
layout <- layout_with_dh(g,set.seed(1))
row.names(layout) <- names(V(g))
colnames(layout) <- c("pos.x","pos.y")
kk <- PathwayExtendData %>%
filter(kegg_pathwayname=="Nicotinate and nicotinamide metabolism")
node.data <- as.data.frame(layout) %>%
tibble::rownames_to_column(var="label") %>%
left_join(diff_gene,by=c("label"="name")) %>%
left_join(diff_meta,by=c("label"="name")) %>%
dplyr::select(label,pos.x,pos.y,logFC.x,logFC.y) %>%
mutate(logFC=ifelse(is.na(logFC.x),logFC.y,logFC.x)) %>%
mutate(type=ifelse(is.na(logFC.x),"metabolite","gene")) %>%
dplyr::select(-logFC.x,-logFC.y) %>%
mutate(label1=ifelse(label %in% gene1$`Gene Name` |label %in% kk$name| type=="metabolite",label,NA))
ab <- node.data %>%
filter(!is.na(label1)) %>%
left_join(gene1,by=c("label1"="Gene Name"))
write.table(ab,"result/Figure4/4E_node-filter.txt",quote=F,row.names = F,sep="\t")
edge.data <- dd %>%
dplyr::rename("fromNode"="X1",
"toNode"="X2") %>%
dplyr::select(fromNode,toNode) %>%
dplyr::left_join(node.data,by=c("fromNode"="label")) %>%
dplyr::rename("from.x"="pos.x","from.y"="pos.y") %>%
dplyr::left_join(node.data,by=c("toNode"="label")) %>%
dplyr::rename("to.x"="pos.x","to.y"="pos.y")
#-------------------------------------------------------------------------------
# Step 2: Core metabolism-based subnetwork
#-------------------------------------------------------------------------------
## node.color
source("raw_data/node.color.R")
source("raw_data/col.key.R")
source("raw_data/colorpanel2.R")
node_meta <- node.data %>%
filter(type=="metabolite") %>%
mutate(logFC=round(logFC,2)) %>%
mutate(logFC=ifelse(logFC < -0.58, -0.58, logFC))
meta_limits=as.numeric(sprintf("%.1f", max(max(node_meta$logFC),abs(min(node_meta$logFC)))))+0.1
meta_color <- node.color(limit=meta_limits,node_meta$logFC,low="#007DDB", mid="gray", high="yellow")
node_meta$color <- meta_color
node_gene <- node.data %>%
filter(type=="gene") %>%
mutate(logFC=round(logFC,2)) %>%
mutate(logFC=ifelse(logFC < -2, -2, logFC)) %>%
mutate(logFC=ifelse(logFC > 2, 2, logFC))
gene_limits=as.numeric(sprintf("%.1f", max(max(node_gene$logFC),abs(min(node_gene$logFC)))))+0.1
gene_color <- node.color(limit=gene_limits,node_gene$logFC,low="#4FBD81",mid="gray",high="#D01910")
node_gene$color <- gene_color
node.data1 <- rbind(node_meta,node_gene)
aa=node.data1$color
names(aa)=node.data1$color
gg <- ggplot()
gg <- gg + geom_segment(mapping = aes(x = from.x, y = from.y, xend = to.x, yend = to.y),
color = "#CCCCCC", size = 0.5, data = edge.data)
gg <- gg + geom_point(mapping = aes(x = pos.x, y = pos.y,color=color,shape=type),
size = 4, data = node.data1)
gg <- gg + scale_size(range = c(0, 6) * 2)
gg <- gg + theme_void()+theme(legend.position="none")
gg <- gg + labs(x = "", y = "")
gg <- gg + ggrepel::geom_text_repel(mapping=aes(x = pos.x, y = pos.y,label=label1),data=node.data1)
gg <- gg + scale_colour_manual(values = aa)+scale_shape_manual(values=c("gene"="square","metabolite"="circle"))
p <- plot.new()
off.sets=col.key(limit=gene_limits,bins=10,cex=0.5,graph.size = c(1,1),off.sets=c(x=0,y=0),
low="#4FBD81",mid="gray",high="#D01910")
off.sets=col.key(limit=meta_limits,bins=10,cex=0.5,low="#007DDB", mid="gray", high="yellow",
off.sets=c(x=0,y=0),graph.size = c(1,0.9))
p <- recordPlot()
dev.off()
p1 <- ggdraw()+
draw_plot(p,0.5,0.5,0.5,0.55)+
draw_plot(gg,0,0,1,1)
ggsave("result/Figure4/4E_node-300-all-B.pdf",p1,width=6,height = 6)
if (0) {
png("result/Figure4/4E_node-300-aa.png",width=5,height = 5,units = 'in', res = 200)
grid::grid.draw(p1)
dev.off()
}
5.6 (F) Discrimination of T-ALL Clusters
The discrimination of two T-ALL clusters (TC1 and TC2) based on SNF method.
library(dplyr)
library(MNet)
library(survival)
library(survminer)
#-------------------------------------------------------------------------------
# Step 1: Load data and set parameters
#-------------------------------------------------------------------------------
dat_filter <- data.table::fread("result/Figure2/1.BMMCvsHC.txt") %>%
as.data.frame() %>%
dplyr::arrange(P.Value) %>%
dplyr::filter(P.Value < 0.5)
dat_filter$P.Value[1:5]
dat_cox <- data.table::fread("result/Figure4/4A_1.cox_metabolite_all_T.txt") %>%
as.data.frame() %>%
dplyr::arrange(p.value) %>%
head(n=120)
dat_cox$p.value[1:5]
dat_filter <- dat_filter %>%
dplyr::filter(name %in% dat_cox$name_raw)
sample_info <- readxl::read_excel("raw_data/sample_clinical_201_info.xlsx") %>%
as.data.frame() %>%
dplyr::filter(!is.na(METc_BM_D0_ID)) %>%
dplyr::filter(!is.na(RNA_ID)) %>%
dplyr::filter(Lineage=="T")
dat_cell <- data.table::fread("raw_data/cell_dat_final_reuslt_v0329.txt") %>%
as.data.frame() %>%
dplyr::filter(label %in% dat_filter$name) %>%
tibble::column_to_rownames("label") %>%
dplyr::select(all_of(sample_info$METc_BM_D0_ID)) %>%
log1p()
names(dat_cell) <- sample_info$Pid
cox_gene <- data.table::fread("result/Figure4/4A_1.cox_gene_all_T.txt") %>%
as.data.frame() %>%
dplyr::arrange(p.value) %>%
head(n=600)
dat_gene <- readRDS("raw_data/20231113_RNA_ALL_VST_coding.rds") %>%
as.data.frame() %>%
dplyr::select(sample_info$RNA_id_raw) %>%
tibble::rownames_to_column(var="label") %>%
dplyr::filter(label %in% cox_gene$name_raw) %>%
tibble::column_to_rownames("label")
names(dat_gene) <- sample_info$Pid
#-------------------------------------------------------------------------------
# Step 2: SNF of discrimination T-ALL clusters
#-------------------------------------------------------------------------------
library(SNFtool)
metabolite_norm = standardNormalization(t(dat_cell))
metgene_norm = standardNormalization(t(dat_gene))
dim(metabolite_norm)
dim(metgene_norm)
write.table(metabolite_norm,"result/Figure4/4F_metabolite_norm_T.txt",quote=F,sep="\t")
write.table(metgene_norm,"result/Figure4/4F_metgene_norm_T.txt",quote=F,sep="\t")
Dist1 = (SNFtool::dist2(as.matrix(metabolite_norm),as.matrix(metabolite_norm)))^(1/2)
Dist2 = (SNFtool::dist2(as.matrix(metgene_norm),as.matrix(metgene_norm)))^(1/2)
W1 = affinityMatrix(Dist1, 10, 0.5)
W2 = affinityMatrix(Dist2, 10, 0.5)
W = SNF(list(W1,W2), 10, 10)
Clusters = spectralClustering(W,K=2)
M_label=cbind(Clusters,sample_info) %>%
dplyr::mutate(Clusters=ifelse(Clusters==1,"TC2",
ifelse(Clusters==2,"TC1",Clusters)))
write.table(M_label,paste0("result/Figure4/4F_cluster_T.txt"),quote=F,row.names = F,sep="\t")
l <- M_label %>%
dplyr::arrange(Clusters)
W_2 <- W %>%
as.data.frame() %>%
dplyr::select(l$Pid) %>%
tibble::rownames_to_column(var="sample") %>%
dplyr::arrange(match(sample,l$Pid)) %>%
tibble::column_to_rownames("sample")
W_2[W_2==0.5] <- 0
col_fun = circlize::colorRamp2(c( 0, 0.03), c( "white","#F48326"))
library(ComplexHeatmap)
p_metabolite <- Heatmap(W_2,height=unit(10,"cm"),name="Sample Data",
cluster_rows = F,cluster_columns = F,
col = col_fun,
#top_annotation =top_annotation,
show_column_names = F,show_row_names = F,
row_names_gp = gpar(fontsize = 3),column_names_gp = gpar(fontsize = 3))
pdf("result/Figure4/4F_SNF_discrimination_complexheatmap_T.pdf",width=5,height = 5)
p_metabolite
dev.off()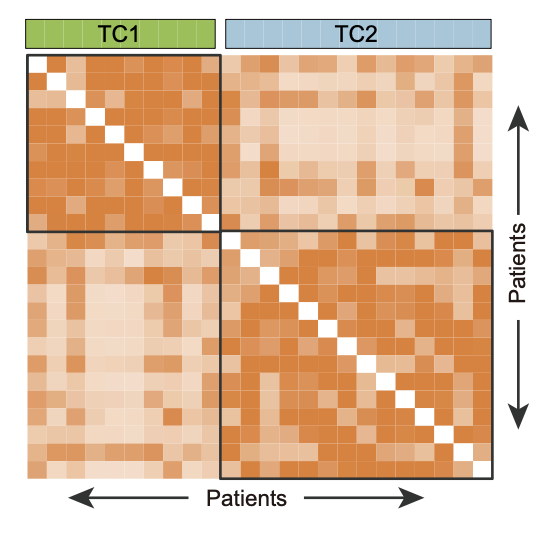
5.7 (G) OS of TC1/2
The association of the two metabolic clusters with clinical OS outcomes in 24 T-ALL patients. The result suggested TC1 was of significantly poorer prognosis than TC2.
library(dplyr)
library(MNet)
library(survival)
library(survminer)
#------------------------------------------------------------------------------
# OS of TC1/2
#------------------------------------------------------------------------------
##### new survival #####
survival_new <- readxl::read_excel("raw_data/Meta_Files.xlsx",skip=1) %>%
as.data.frame() %>%
dplyr::select(PID,OS_stat,`OS (days)`) %>%
dplyr::rename("OS"="OS (days)")
clinical_survival <- data.table::fread(paste0("result/Figure4/4F_cluster_T.txt")) %>%
as.data.frame() %>%
dplyr::select(-OS_stat,-OS,-EFS_stat,-EFS,-RFS_stat,-RFS) %>%
left_join(survival_new,by=c("Pid"="PID"))
surv_object <- Surv(time = clinical_survival$OS/30, event = clinical_survival$OS_stat)
fit_cluster <- survfit(surv_object ~ Clusters, data = clinical_survival)
p1 <- survminer::ggsurvplot(fit_cluster, data = clinical_survival,
title="OS",
palette=c("#9BD53F","#A6CEE3"),
tables.theme = theme_void(),risk.table = TRUE,
risk.table.fontsize=6,
break.time.by = 12,
risk.table.y.text = FALSE,
tables.height = 0.15,risk.table.col = "black",
pval = TRUE,
pval.method = F,pval.coord=c(0.5,0.1),pval.size=6,
font.tickslab=c(14,"plain","black"),
xlab="Time (Months) ",ylab="OS Survival")
pdf(paste0("result/Figure4/4G_OS_T.pdf"),onefile=FALSE)
p1
dev.off()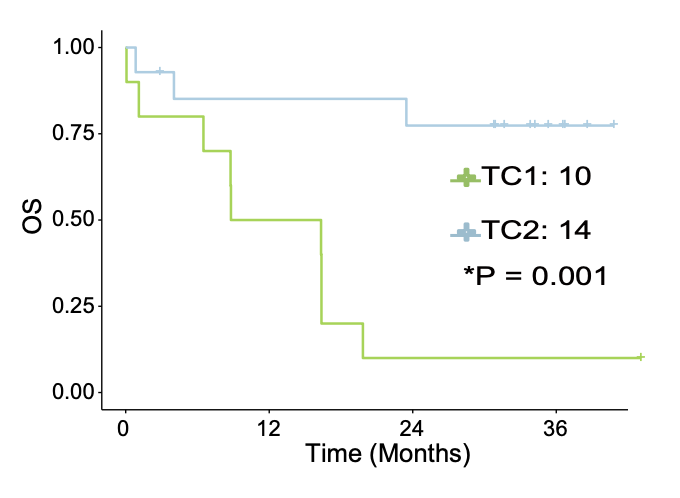
5.8 (H) Subnetwork between TC2 and TC1
A core metabolism-based subnetwork that best explains the difference between TC2 and TC1. The highlighted labels correspond to pathways in the TCA and OXPHOS. PC refers to a group of phosphatidylcholines without specific KEGG IDs.
library(dplyr)
library(ggplot2)
library(igraph)
library(MNet)
library(graphics)
library(cowplot)
#-------------------------------------------------------------------------------
# Step 1: Read data and set parameters
#-------------------------------------------------------------------------------
rm(list = ls())
name_gene <- data.table::fread("result/Figure4/4A_1.cox_gene_all_T.txt") %>%
as.data.frame()
diff_gene <- data.table::fread("result/Figure4/S6J_gene_T.txt") %>%
as.data.frame() %>%
filter(name %in% name_gene$name_raw)
kid <- openxlsx::read.xlsx("raw_data/cell_metabolite_info_all_v1109.xlsx") %>%
as.data.frame() %>%
dplyr::select(refmet_name,KEGG) %>%
mutate(KEGG=ifelse(KEGG=="None",refmet_name,KEGG))
diff_meta <- data.table::fread("result/Figure4/S6J_metabolite_T.txt") %>%
as.data.frame() %>%
inner_join(kid,by=c("name"="refmet_name")) %>%
dplyr::select(-name) %>%
dplyr::rename("name"="KEGG") %>%
arrange(P.Value) %>%
distinct(name,.keep_all = T)
names(diff_meta)[4] <- "p_value"
names(diff_gene)[4] <- "p_value"
dd <- data.table::fread("raw_data/subnetwork_important_edge-300_T.txt") %>%
as.data.frame()
set.seed(2)
g <- graph_from_data_frame(dd)
layout <- layout_with_dh(g,set.seed(1))
row.names(layout) <- names(V(g))
colnames(layout) <- c("pos.x","pos.y")
kk <- PathwayExtendData %>%
filter(kegg_pathwayname %in% c("Citrate cycle (TCA cycle)","Oxidative phosphorylation"))
node.data <- as.data.frame(layout) %>%
tibble::rownames_to_column(var="label") %>%
left_join(diff_gene,by=c("label"="name")) %>%
left_join(diff_meta,by=c("label"="name")) %>%
dplyr::select(label,pos.x,pos.y,logFC.x,logFC.y) %>%
mutate(logFC=ifelse(is.na(logFC.x),logFC.y,logFC.x)) %>%
mutate(type=ifelse(is.na(logFC.x),"metabolite","gene")) %>%
dplyr::select(-logFC.x,-logFC.y) %>%
mutate(label1=ifelse(label %in% kk$name,label,NA))
edge.data <- dd %>%
dplyr::rename("fromNode"="X1",
"toNode"="X2") %>%
dplyr::select(fromNode,toNode) %>%
dplyr::left_join(node.data,by=c("fromNode"="label")) %>%
dplyr::rename("from.x"="pos.x","from.y"="pos.y") %>%
dplyr::left_join(node.data,by=c("toNode"="label")) %>%
dplyr::rename("to.x"="pos.x","to.y"="pos.y")
## node.color
source("raw_data/node.color.R")
source("raw_data/col.key.R")
source("raw_data/colorpanel2.R")
node_meta <- node.data %>%
filter(type=="metabolite") %>%
mutate(logFC=round(logFC,2)) %>%
mutate(logFC=ifelse(logFC < -0.58, -0.58, logFC))
meta_limits=as.numeric(sprintf("%.1f", max(max(node_meta$logFC),abs(min(node_meta$logFC)))))+0.1
meta_color <- node.color(limit=meta_limits,node_meta$logFC,low="#007DDB", mid="gray", high="yellow")
node_meta$color <- meta_color
node_gene <- node.data %>%
filter(type=="gene") %>%
mutate(logFC=round(logFC,2)) %>%
mutate(logFC=ifelse(logFC < -2, -2, logFC)) %>%
mutate(logFC=ifelse(logFC > 2, 2, logFC))
gene_limits=as.numeric(sprintf("%.1f", max(max(node_gene$logFC),abs(min(node_gene$logFC)))))+0.1
gene_color <- node.color(limit=gene_limits,node_gene$logFC,low="#4FBD81",mid="gray",high="#D01910")
node_gene$color <- gene_color
node.data1 <- rbind(node_meta,node_gene)
aa=node.data1$color
names(aa)=node.data1$color
gg <- ggplot()
gg <- gg + geom_segment(mapping = aes(x = from.x, y = from.y, xend = to.x, yend = to.y),
color = "#CCCCCC", size = 0.5, data = edge.data)
gg <- gg + geom_point(mapping = aes(x = pos.x, y = pos.y,color=color,shape=type),
size = 4, data = node.data1)
gg <- gg + scale_size(range = c(0, 6) * 2)
gg <- gg + theme_void()+theme(legend.position="none")
gg <- gg + labs(x = "", y = "")
gg <- gg + ggrepel::geom_text_repel(mapping=aes(x = pos.x, y = pos.y,label=label1),data=node.data1)
gg <- gg + scale_colour_manual(values = aa)+scale_shape_manual(values=c("gene"="square","metabolite"="circle"))
pdf("result/Figure4/4H_node-200-1_T.pdf")
p <- plot.new()
off.sets=col.key(limit=gene_limits,bins=10,cex=0.5,graph.size = c(1,1),off.sets=c(x=0,y=0),
low="#4FBD81",mid="gray",high="#D01910")
off.sets=col.key(limit=meta_limits,bins=10,cex=0.5,low="#007DDB", mid="gray", high="yellow",
off.sets=c(x=0,y=0),graph.size = c(1,0.9))
p <- recordPlot()
dev.off()
if (0) {
p1 <- ggdraw()+
draw_plot(p,0.5,0.5,0.5,0.55)+
draw_plot(gg,0,0,1,1)
ggsave("result/Figure4/4H_node-300-all.pdf",p1,width=6,height = 6)
}
ggsave("result/Figure4/4H_node-300-2_T.pdf",gg,width=6,height = 6)
if (0) {
png("result/Figure4/4H_node-300-aa.png",width=5,height = 5,units = 'in', res = 200)
grid::grid.draw(p1)
dev.off()
}
5.9 (S5 A) Clusters of BCP-ALL
The relative abundance of input metabolites and expression level of metabolic genes in the three metabolic clusters in BCP-ALL: BC1 (N=20), BC2 (N=35), and BC3 (N=20). Each column represents a sample and each row represents a metabolite (upper panel) or gene included (lower panel).
#------------------------------------------------------------------------------
# Step 1: Heatmap
#------------------------------------------------------------------------------
library(dplyr)
library(ComplexHeatmap)
sample_info <- data.table::fread("result/Figure4/4B_cluster_B.txt") %>%
as.data.frame()
metabolite_norm = data.table::fread("result/Figure4/4B_metabolite_norm_B.txt") %>%
as.data.frame() %>%
tibble::column_to_rownames("V1")
metgene_norm <- data.table::fread("result/Figure4/4B_metgene_norm_B.txt") %>%
as.data.frame() %>%
tibble::column_to_rownames("V1")
top_annotation = HeatmapAnnotation(Subtype=sample_info$Clusters,
col=list(Subtype=c("BC1"="#9BD53F","BC2"="#A6CEE3","BC3"="#EA644C")),
show_annotation_name = c(T,T),
border = c(T,T) ,
simple_anno_size = unit(4, "mm"),gap = unit(1, "mm"))
col_fun = circlize::colorRamp2(c(-1, 0, 1), c("#00599F", "white", "#D01910"))
p_metabolite <- Heatmap(t(metabolite_norm),height=unit(10,"cm"),name="Metabolite Data",
cluster_row_slices = F,clustering_method_rows = "ward.D",
clustering_method_columns = "ward.D2",
column_split = sample_info$Clusters,
col = col_fun,
top_annotation =top_annotation,
show_column_names = F,show_row_names = F,
row_names_gp = gpar(fontsize = 3),
column_names_gp = gpar(fontsize = 3))
col_fun = circlize::colorRamp2(c(-1, 0, 1), c("#00599F", "white", "#FF7F00"))
p_gene <- Heatmap(t(metgene_norm),height=unit(10,"cm"),name="Gene Data",
cluster_row_slices = F,clustering_method_rows = "ward.D",
clustering_method_columns = "ward.D2",
column_split = sample_info$Clusters,
col = col_fun,
show_column_names = F,show_row_names = F,
row_names_gp = gpar(fontsize = 3),
column_names_gp = gpar(fontsize = 3))
pdf(paste0("result/Figure4/S5A_heatmap_B.pdf"),width=8,height = 10)
p_metabolite %v% p_gene
dev.off()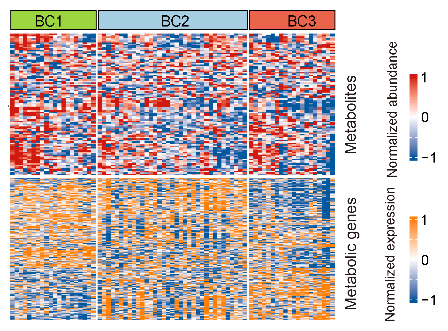
5.10 (S5 G) Waterfall plot of BC1/2/3
The associations of metabolic clusters with clinical variables and genetic alterations, including major mutations and fusions in BCP-ALL. Note: Among all fusions, only BCR::ABL1 and TCF3::PBX1 were significantly enriched in BC1/2 and BC3, respectively. P-value was calculated by Pearson’s chi-square test or Fisher’s exact test.
library(dplyr)
library(ComplexHeatmap)
#-------------------------------------------------------------------------------
# Step 1: Load data and set parameters
#-------------------------------------------------------------------------------
clinical_survival <- readxl::read_excel("raw_data/Meta_Files.xlsx",skip=1) %>%
as.data.frame() %>%
dplyr::rename("OS"="OS (days)","EFS"="EFS (days)","RFS"="RFS (days)","ifHSCT"="HSCT_stat",
"WBC"="WBC (10⁹/L)","Age"="Age (yrs)","Pid"="PID","RNA_id_raw"="RNA_ID_raw") %>%
dplyr::select(Pid,RNA_id_raw,Age,Sex,WBC,ifHSCT,OS_stat,D28_status) %>%
#dplyr::mutate(Age=ifelse(Age < 40,"< 40",">= 40")) %>%
dplyr::mutate(WBC_type= ifelse(WBC>30, "> 30",
ifelse(WBC<=30,"<= 30",NA))) %>%
dplyr::mutate(Outcome=ifelse(OS_stat==1,"Dead",
ifelse(OS_stat==0,"Alive",OS_stat))) %>%
dplyr::mutate(ifHSCT=ifelse(ifHSCT==0,"no",
ifelse(ifHSCT==1,"yes",ifHSCT)))
sample_info_raw <- data.table::fread("raw_data/1.cluster_B.txt") %>%
as.data.frame() %>%
dplyr::select(bianhao,Clusters)
sample_info <- data.table::fread("result/Figure4/4B_cluster_B.txt") %>%
as.data.frame() %>%
dplyr::select(Clusters,Pid) %>%
dplyr::mutate(bianhao=gsub("RJ-","A",Pid)) %>%
dplyr::arrange(match(bianhao,sample_info_raw$bianhao)) %>%
dplyr::left_join(clinical_survival,by="Pid") %>%
dplyr::rename("RNA_id"="RNA_id_raw")
#-------------------------------------------------------------------------------
# Step 2: Mutations
#-------------------------------------------------------------------------------
q12 <- c("#1F78B4","#E31A1C","black")
names(q12) <- unique(sample_info$Clusters)
top_annotation = HeatmapAnnotation(
Subtype=sample_info$Clusters,
#Age=sample_info$Age,
ifHSCT=sample_info$ifHSCT,
WBC=sample_info$WBC_type,
Outcome=sample_info$Outcome,
col=list(Subtype=c("BC1"="#9BD53F","BC2"="#A6CEE3","BC3"="#EA644C"),
Age=c("< 40"="#B2DF8A",">= 40"="gray"),
ifHSCT=c("yes"="#CAB2D6","no"="gray"),
WBC=c("> 30"="#FDBF6F","<= 30"="gray"),
Outcome=c("Dead"="black","Alive"="gray")),
show_annotation_name = c(T,T),
border = c(T,T,T,T,T,T) ,
simple_anno_size = unit(8, "mm"),gap = unit(1.5, "mm"))
mutation_B <- data.table::fread("raw_data/mutation_B_v1114_all.txt") %>%
as.data.frame() %>%
dplyr::select(V2,V17,V19) %>%
dplyr::rename("ID"="V2","Gene.refGene"="V17","ExonicFunc.refGene"="V19") %>%
dplyr::select(ID,`Gene.refGene`,`ExonicFunc.refGene`) %>%
dplyr::mutate(`ExonicFunc.refGene`=
ifelse(`ExonicFunc.refGene` %in% c("frameshift deletion","frameshift insertion","frameshift substitution"),"Frameshift",
ifelse(`ExonicFunc.refGene` %in% c("nonframeshift deletion","nonframeshift insertion","nonframeshift substitution"),"Nonframeshift",
ifelse(`ExonicFunc.refGene` %in% ".","Splice",
ifelse(`ExonicFunc.refGene` %in% "nonsynonymous SNV","Missense",
ifelse(`ExonicFunc.refGene`=="startloss","Startloss",
ifelse(`ExonicFunc.refGene`=="stopgain","Stopgain",
ifelse(`ExonicFunc.refGene`=="synonymous SNV","Silent",`ExonicFunc.refGene`)))))))) %>%
unique() %>%
dplyr::filter(ID %in% sample_info$RNA_id) %>%
dplyr::mutate(`Gene.refGene` = ifelse(`Gene.refGene` %in% c("IDH1","IDH2"),"IDH1/2",`Gene.refGene`))
mutation <- rbind(mutation_B)
hit_num <- mutation %>%
dplyr::group_by(ID,`Gene.refGene`) %>%
dplyr::summarise(n=n()) %>%
dplyr::filter(n>1)
mutation <- mutation %>%
dplyr::left_join(hit_num,by=c("ID","Gene.refGene")) %>%
dplyr::mutate(`ExonicFunc.refGene`=ifelse(!is.na(n),"Multiple",`ExonicFunc.refGene`)) %>%
dplyr::select(-n) %>%
unique() %>%
dplyr::rename("RNA_id"="ID") %>%
dplyr::filter(RNA_id %in% sample_info$RNA_id)
mutation_gene_filter <- mutation %>%
dplyr::select(RNA_id,Gene.refGene) %>%
unique() %>%
dplyr::group_by(`Gene.refGene`) %>%
dplyr::summarise(n=n()) %>%
dplyr::filter(n>0)
mutation_sort <- rev(names(sort(table(mutation %>% filter(`Gene.refGene` %in% mutation_gene_filter$Gene.refGene) %>% pull(Gene.refGene)))))
mutation <- mutation %>%
dplyr::filter(`Gene.refGene` %in% mutation_gene_filter$Gene.refGene) %>%
as.data.frame() %>%
dplyr::distinct(RNA_id,Gene.refGene,.keep_all = T)
sample_info_filter <- sample_info %>%
dplyr::select(bianhao,RNA_id)
mutation_dcast <- reshape2::dcast(mutation,RNA_id ~ `Gene.refGene`) %>%
dplyr::right_join(sample_info_filter,by="RNA_id") %>%
dplyr::select(-RNA_id) %>%
tibble::column_to_rownames("bianhao") %>%
dplyr::select(all_of(mutation_sort))
mutation_dcast[is.na(mutation_dcast)] <- 0
mutation_dcast[mutation_dcast=="Missense"] <- 1
mutation_dcast[mutation_dcast=="Frameshift"] <- 2
mutation_dcast[mutation_dcast=="Nonframeshift"] <- 3
mutation_dcast[mutation_dcast=="Silent"] <- 4
mutation_dcast[mutation_dcast=="Splice"] <- 5
mutation_dcast[mutation_dcast=="Stopgain"] <- 7
mutation_dcast[mutation_dcast=="Multiple"] <- 8
mutation_dcast1 <- mutation_dcast %>%
dplyr::mutate_if(is.character,as.numeric) %>%
rowwise() %>%
dplyr::mutate(Signaling=sum(NRAS,KRAS,FLT3,STAT5B,NF1,MYC)) %>%
dplyr::mutate(Transcription=sum(PAX5,TP53,RUNX1,IKZF1,ETV6,MGA)) %>%
dplyr::mutate(Epigenetic=sum(KMT2D,SETD2,NCOR2,EP300,ARID1A,ARID1B,SETD1B,`IDH1/2`)) %>%
dplyr::mutate(Others=sum(SACS)) %>%
dplyr::mutate(Signaling=ifelse(Signaling==0,0,9)) %>%
dplyr::mutate(Transcription=ifelse(Transcription==0,0,9)) %>%
dplyr::mutate(Epigenetic=ifelse(Epigenetic==0,0,9)) %>%
dplyr::mutate(Others=ifelse(Others==0,0,9)) %>%
dplyr::select(NRAS,FLT3,STAT5B,NF1,KRAS,MYC,Signaling,
PAX5,TP53,RUNX1,IKZF1,ETV6,MGA,Transcription,
KMT2D,SETD2,NCOR2,EP300,ARID1A,ARID1B,SETD1B,`IDH1/2`,Epigenetic,
SACS,Others) %>%
as.data.frame()
rownames(mutation_dcast1) <- rownames(mutation_dcast)
mutation_dcast <- mutation_dcast1
mutation_dcast_temp <- mutation_dcast
mutation_dcast_temp[mutation_dcast_temp!=0] <- 1
num=colSums(mutation_dcast_temp)
names(mutation_dcast) <- paste0(names(mutation_dcast)," (n=",num,")")
d_mutation <- t(mutation_dcast) %>%
as.data.frame() %>%
dplyr::select(sample_info$bianhao)
mutation_annotation <- Heatmap(d_mutation,height=unit(12,"cm"),rect_gp = gpar(col = "gray", lwd = 2),
col=c("#EEEEEE","#3987CC","#DB3D3D","#663FFB","#FF7F0E","#CC66FF","#8B564C","#339933","blue"),
cluster_columns = F,cluster_rows = F,border = TRUE,column_title = NULL,
column_split =sample_info$Clusters,
show_column_names = TRUE,
top_annotation=top_annotation,
row_names_gp = gpar(fontsize = 10),column_names_gp=gpar(fontsize=6))
#-------------------------------------------------------------------------------
# Step 3: Fusions
#-------------------------------------------------------------------------------
fusion_B <- readxl::read_excel("raw_data/fusion_B_v0803.xlsx",skip=1) %>%
as.data.frame() %>%
dplyr::filter(RNA_id %in% sample_info$RNA_id) %>%
dplyr::select(RNA_id,fusion_group) %>%
dplyr::mutate(fusion_group=ifelse(fusion_group %in% c("PAX5-ETV6","PAX5-CPLX1"),"PAX5 fusion",
fusion_group)) %>%
dplyr::mutate(fusion_group=ifelse(fusion_group=="BCR-ABL1;IKZF1-IGH","BCR-ABL1",fusion_group)) %>%
dplyr::left_join(sample_info,by="RNA_id") %>%
dplyr::filter(fusion_group %in% c("BCL2/MYC","BCR-ABL1","DUX4 rearrangement","ETV6-RUNX1","KMT2A rearrangement","PAX5 fusion","TCF3-PBX1","ZNF384 fusion")) %>%
dplyr::select(RNA_id,fusion_group) %>%
dplyr::filter(!is.na(fusion_group))
fusion <- rbind(fusion_B)
clinical_fusion_dcast <- reshape2::dcast(fusion,RNA_id ~ fusion_group) %>%
dplyr::right_join(sample_info_filter,by="RNA_id") %>%
tibble::column_to_rownames("bianhao") %>%
dplyr::select(-RNA_id)
clinical_fusion_dcast[is.na(clinical_fusion_dcast)] <- 0
clinical_fusion_dcast[clinical_fusion_dcast != 0] <- 1
d_fusion <- t(clinical_fusion_dcast) %>%
as.data.frame() %>%
dplyr::select(sample_info$bianhao) %>%
tibble::rownames_to_column(var="fusion") %>%
dplyr::arrange(match(fusion,c("DUX4 rearrangement","ZNF384 fusion","TCF3-PBX1","KMT2A rearrangement","ETV6-RUNX1","PAX5-ETV6","PAX5 fusion","BCL2/MYC"))) %>%
tibble::column_to_rownames("fusion")
fusion_annotation <- Heatmap(d_fusion,rect_gp = gpar(col = "gray", lwd = 2),
col=c("#EEEEEE","#444444"),
height=unit(4,"cm"),border = TRUE,
column_split =sample_info$Clusters,
cluster_columns = F,cluster_rows = F,
column_title = NULL,
row_names_gp = gpar(fontsize = 10),
column_names_gp=gpar(fontsize=6))
pdf(paste0("result/Figure4/S5G_oncoplot_B.pdf"),width=10,height = 10)
mutation_annotation %v% fusion_annotation
dev.off()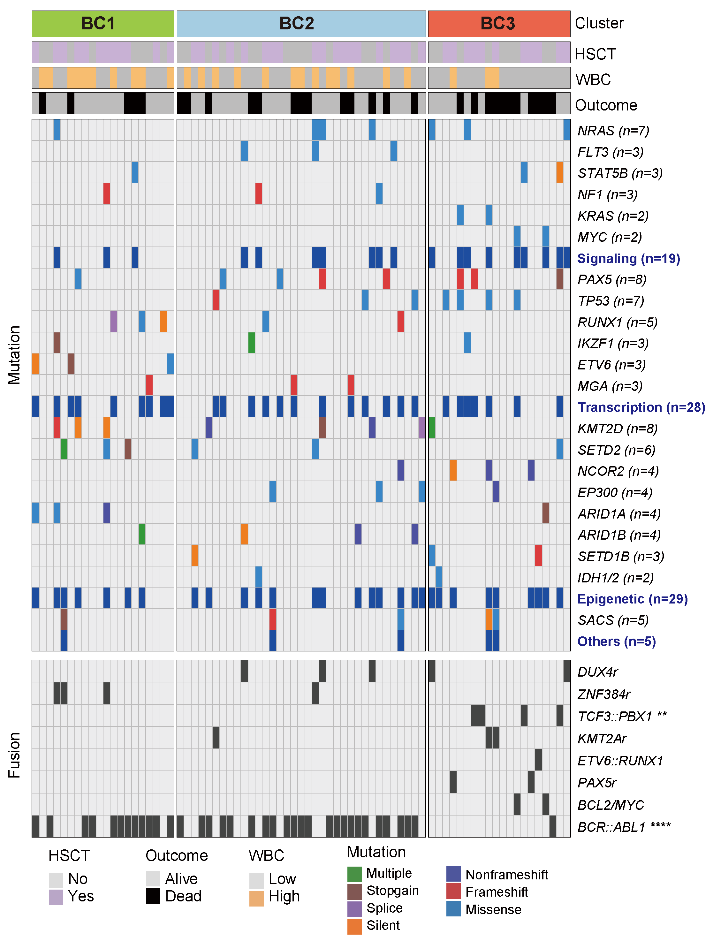
5.11 (S5 H) DA score of BC3 cluster
The DA score reveals that most of the disturbed metabolism pathways are significantly enriched in the BC3 cluster.
library(ggplot2)
library(MNet)
library(dplyr)
#------------------------------------------------------------------------------
# Stpe 1: mlimma of BCP-ALL clusters
#------------------------------------------------------------------------------
cluster <- data.table::fread("result/Figure4/4B_cluster_B.txt") %>%
as.data.frame()
dat <- data.table::fread("raw_data/cell_dat_final_reuslt_v0329.txt") %>%
dplyr::select(label,all_of(cluster$METc_BM_D0_ID)) %>%
tibble::column_to_rownames("label")
group <- cluster$Clusters
group[which(group %in% c("BC3"))] <- "tumor"
group[which(group %in% c("BC1","BC2"))] <- "normal"
table(group)
metabolite_all <- mlimma(log1p(dat),group)
write.table(metabolite_all,"result/Figure4/S5H_metabolite_B.txt",quote=F,row.names=F,sep="\t")
cluster <- data.table::fread("result/Figure4/4B_cluster_B.txt") %>%
as.data.frame()
dat <- readRDS("raw_data/20231113_RNA_ALL_VST_coding.rds") %>%
as.data.frame() %>%
dplyr::select(all_of(cluster$RNA_id_raw))
group <- cluster$Clusters
group[which(group %in% c("BC3"))] <- "tumor"
group[which(group %in% c("BC1","BC2"))] <- "normal"
table(group)
gene_all <- mlimma(dat,group)
write.table(gene_all,"result/Figure4/S5H_gene_B.txt",quote=F,row.names=F,sep="\t")
#------------------------------------------------------------------------------
# Step 2: DA-score of metabolites
#------------------------------------------------------------------------------
kegg_id <- readxl::read_excel("raw_data/cell_metabolite_info_all_v1109.xlsx") %>%
as.data.frame() %>%
dplyr::select(refmet_name,KEGG) %>%
tidyr::separate_rows(KEGG,sep=";") %>%
dplyr::filter(KEGG!="NA") %>%
dplyr::filter(KEGG != "None") %>%
as.data.frame()
dat <- data.table::fread("result/Figure4/S5H_metabolite_B.txt") %>%
dplyr::arrange(desc(logFC)) %>%
dplyr::inner_join(kegg_id,by=c("name"="refmet_name")) %>%
dplyr::distinct(KEGG,.keep_all = TRUE)
dat_increase <- dat %>%
dplyr::filter(logFC > 0) %>%
dplyr::filter(P.Value < 0.05)
dat_decrease <- dat %>%
dplyr::filter(logFC < 0) %>%
dplyr::filter(P.Value < 0.05)
DA_result <- DAscore(dat_increase$KEGG,dat_decrease$KEGG,dat$KEGG,min_measured_num = 3,out="metabolite")
write.table(DA_result$result,"result/Figure4/S5H_DAscore_metabolite_B.txt",quote=F,row.names=F,sep="\t")
#------------------------------------------------------------------------------
# Step 3: DA-score of genes
#------------------------------------------------------------------------------
dat <- data.table::fread("result/Figure4/S5H_gene_B.txt")
dat_increase <- dat %>%
dplyr::filter(logFC > 0) %>%
dplyr::filter(P.Value < 0.05)
dat_decrease <- dat %>%
dplyr::filter(logFC < 0) %>%
dplyr::filter(P.Value < 0.05)
DA_result <- DAscore(dat_increase$name,dat_decrease$name,dat$name,min_measured_num = 10,out="gene")
write.table(DA_result$result,"result/Figure4/S5H_DAscore_gene_B.txt",quote=F,row.names=F,sep="\t")
#------------------------------------------------------------------------------
# Step 4: DA-score of metabolites+genes
#------------------------------------------------------------------------------
gene <- data.table::fread("result/Figure4/S5H_DAscore_gene_B.txt") %>%
as.data.frame() %>%
dplyr::filter(Measured_members_num >= 10) %>%
dplyr::filter(abs(DA_score) > 0.05)
metabolite <- data.table::fread("result/Figure4/S5H_DAscore_metabolite_B.txt") %>%
as.data.frame() %>%
dplyr::filter(Measured_members_num >= 3) %>%
dplyr::filter(DA_score != 0)
all_1 <- gene %>%
dplyr::inner_join(metabolite,by="Pathway") %>%
dplyr::filter(DA_score.x >0 & DA_score.y >0)
all_2 <- gene %>%
dplyr::inner_join(metabolite,by="Pathway") %>%
dplyr::filter(DA_score.x < 0 & DA_score.y < 0)
path <- c(all_1$Pathway,all_2$Pathway,"Nicotinate and nicotinamide metabolism")
metabolite_filter <- metabolite %>%
dplyr::filter(Pathway %in% path) %>%
dplyr::filter(Measured_members_num >= 3) %>%
dplyr::filter(abs(DA_score) > 0.05) %>%
dplyr::arrange(`Pathway Category`) %>%
dplyr::mutate(Pathway=factor(Pathway,levels = Pathway))
colp <- c("Amino acid metabolism" ="#1B9E77",
"Carbohydrate metabolism"="#D95F02",
"Glycan biosynthesis and metabolism"="#1F78B4",
"Metabolism of cofactors and vitamins"="#7570B3",
"Metabolism of terpenoids and polyketides"="#BC80BD",
"Metabolism of other amino acids"="#8DD3C7",
"Energy metabolism"="#E7298A",
"Lipid metabolism"="#66A61E",
"Nucleotide metabolism"="#E6AB02",
"Biosynthesis of other secondary metabolites"="#A6761D",
"Xenobiotics biodegradation and metabolism"="#666666")
p <- ggplot2::ggplot(metabolite_filter)+
ggplot2::geom_point(ggplot2::aes(x=Pathway,y=DA_score,size=log2(Measured_members_num),color=`Pathway Category`))+
ggplot2::geom_pointrange(ggplot2::aes(x=Pathway,y=DA_score,ymin=0,ymax=DA_score,color=`Pathway Category`))+
scale_color_manual(values=colp)+
ggplot2::coord_flip()+
ggplot2::xlab(NULL)+
ggplot2::theme_bw()
ggsave("result/Figure4/S5H_DAscore_metabolite_B_filter.pdf",p,width=8,height = 5)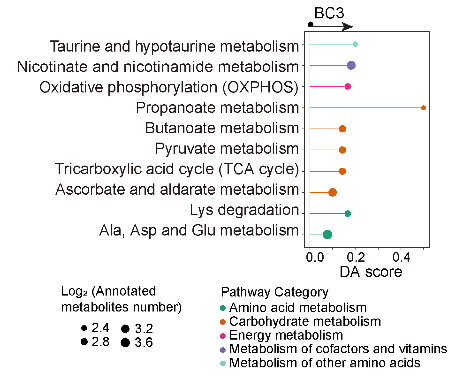
5.12 (S5 I-J) GSEA between BC3 and BC1/2
I. GSEA unveils pathways significantly enriched in BC3, mirroring the findings of the metabolome analyses.
J. GSEA unveils that cholesterol biosynthesis was significantly enriched in BC3.
#------------------------------------------------------------------------------
# Step 1: KEGG citrate TCA cycle
#------------------------------------------------------------------------------
library(dplyr)
library(ggplot2)
## Load data and set parameters
gene_all <- data.table::fread("result/Figure4/S5H_gene_B.txt") %>%
as.data.frame()
a <- readLines("raw_data/tca.txt")
gmt <- strsplit(a, "\t")
names(gmt) <- vapply(gmt, function(y) y[1], character(1))
gmt1 <- lapply(gmt, "[", -c(1:2))
a_temp <- gene_all$logFC
names(a_temp) <- gene_all$name
Ranks_all <- sort(a_temp,decreasing =TRUE)
gseaParam = 0.5
minSize=5
ticksSize = 0.2
Ranks_all = sort(a_temp,decreasing =TRUE)
fgseaRes_all <- fgsea::fgsea(gmt1, sort(a_temp,decreasing =TRUE),minSize = minSize,nPermSimple=20000,gseaParam=0.5)
pval <- fgseaRes_all %>%
dplyr::pull(pval) %>%
round(3)
nes <- fgseaRes_all %>%
dplyr::pull(NES) %>%
round(3)
pathway <- gmt1[[1]]
stats <- Ranks_all
rnk <- rank(-stats)
ord <- order(rnk)
statsAdj <- stats[ord]
statsAdj <- sign(statsAdj) * (abs(statsAdj)^gseaParam)
statsAdj <- statsAdj/max(abs(statsAdj))
pathway <- unname(as.vector(na.omit(match(pathway, names(statsAdj)))))
pathway <- sort(pathway)
gseaRes <- fgsea::calcGseaStat(statsAdj, selectedStats = pathway,
returnAllExtremes = TRUE)
bottoms <- gseaRes$bottoms
tops <- gseaRes$tops
n <- length(statsAdj)
xs <- as.vector(rbind(pathway - 1, pathway))
ys <- as.vector(rbind(bottoms, tops))
toPlot <- data.frame(x = c(0, xs, n + 1), y = c(0, ys, 0))
diff <- (max(tops) - min(bottoms))/8
x = y = NULL
## Visualization
g1 <- ggplot2::ggplot(toPlot, aes(x = x, y = y)) +
ggplot2::geom_point(color = "green", size = 0.1) +
ggplot2::geom_hline(yintercept = 0, colour = "gray") +
geom_line(color = "green") +
ggplot2::geom_line(color = "green") +
theme_bw() +
ggplot2::theme(panel.grid.minor = ggplot2::element_blank(),
axis.text.x=ggplot2::element_blank(),axis.ticks.x=ggplot2::element_blank()) +
ggplot2::labs(x = NULL, y = "enrichment score",title=)+
ggplot2::scale_x_continuous(expand = c(0, 0),limits = c(0,length(statsAdj)+1))+
ggplot2::theme(plot.margin = unit(c(5,5,0,5), "mm"))
g2 <- ggplot2::ggplot(toPlot, aes(x = x, y = y))+
ggplot2::geom_segment(data = data.frame(x = pathway),
mapping = aes(x = x,
y = -1, xend = x, yend = 1),
size = ticksSize,color="red") +
ggplot2::scale_x_continuous(expand = c(0, 0),
limits = c(0,length(statsAdj)+1))+
theme_bw()+
ggplot2::theme(panel.grid = element_blank(),
axis.text.x=element_blank(),axis.ticks.x=element_blank(),
axis.text.y=element_blank(),axis.ticks.y=element_blank())+
ggplot2::labs(x=NULL,y=NULL)+
ggplot2::theme(plot.margin = unit(c(0,5,0,5), "mm"))
dat <- data.frame(name=1:length(statsAdj),value=statsAdj)
g3 <- ggplot2::ggplot(dat,aes(name,value))+
ggplot2::geom_bar(stat="identity",fill="gray")+
ggplot2::theme_bw()+
ggplot2::theme(panel.grid = element_blank())+
ggplot2::labs(x=NULL)+
ggplot2::scale_x_continuous(expand = c(0, 0),limits=c(0,length(statsAdj)+1))+
ggplot2::theme(plot.margin = unit(c(0,5,5,5), "mm"))
p1 <- cowplot::plot_grid(plotlist = list(g1,g2,g3),nrow=3,
rel_heights = c(1,0.2,0.5),scale=1,
align ="v",labels=paste0("TCA Cycle"," pval=",pval,";NES=",nes),label_size = 8)
ggsave("result/Figure4/S5I_GSEA_B_TCA.pdf",p1,width=4,height = 4)
#------------------------------------------------------------------------------
# Step 2: Mitochondrial gene expression
#------------------------------------------------------------------------------
library(dplyr)
library(ggplot2)
## Load data and set parameters
gene_all <- data.table::fread("result/Figure4/S5H_gene_B.txt") %>%
as.data.frame()
a <- readLines("raw_data/MITOCHONDRIAL.txt")
gmt <- strsplit(a, "\t")
names(gmt) <- vapply(gmt, function(y) y[1], character(1))
gmt1 <- lapply(gmt, "[", -c(1:2))
a_temp <- gene_all$logFC
names(a_temp) <- gene_all$name
Ranks_all <- sort(a_temp,decreasing =TRUE)
gseaParam = 0.5
minSize=5
ticksSize = 0.2
Ranks_all = sort(a_temp,decreasing =TRUE)
fgseaRes_all <- fgsea::fgsea(gmt1, sort(a_temp,decreasing =TRUE),minSize = minSize,nPermSimple=20000,gseaParam=0.5)
pval <- fgseaRes_all %>%
dplyr::pull(pval) %>%
round(3)
nes <- fgseaRes_all %>%
dplyr::pull(NES) %>%
round(3)
pathway <- gmt1[[1]]
stats <- Ranks_all
rnk <- rank(-stats)
ord <- order(rnk)
statsAdj <- stats[ord]
statsAdj <- sign(statsAdj) * (abs(statsAdj)^gseaParam)
statsAdj <- statsAdj/max(abs(statsAdj))
pathway <- unname(as.vector(na.omit(match(pathway, names(statsAdj)))))
pathway <- sort(pathway)
gseaRes <- fgsea::calcGseaStat(statsAdj, selectedStats = pathway,
returnAllExtremes = TRUE)
bottoms <- gseaRes$bottoms
tops <- gseaRes$tops
n <- length(statsAdj)
xs <- as.vector(rbind(pathway - 1, pathway))
ys <- as.vector(rbind(bottoms, tops))
toPlot <- data.frame(x = c(0, xs, n + 1), y = c(0, ys, 0))
diff <- (max(tops) - min(bottoms))/8
x = y = NULL
## Visualization
g1 <- ggplot2::ggplot(toPlot, aes(x = x, y = y)) +
ggplot2::geom_point(color = "green", size = 0.1) +
ggplot2::geom_hline(yintercept = 0,
colour = "gray") + geom_line(color = "green") +
ggplot2::geom_line(color = "green") + theme_bw() +
ggplot2::theme(panel.grid.minor = ggplot2::element_blank(),
axis.text.x=ggplot2::element_blank(),axis.ticks.x=ggplot2::element_blank()) +
ggplot2::labs(x = NULL, y = "enrichment score",title=)+
ggplot2::scale_x_continuous(expand = c(0, 0),limits = c(0,length(statsAdj)+1))+
ggplot2::theme(plot.margin = unit(c(5,5,0,5), "mm"))
g2 <- ggplot2::ggplot(toPlot, aes(x = x, y = y))+
ggplot2::geom_segment(data = data.frame(x = pathway),
mapping = aes(x = x,
y = -1, xend = x, yend = 1),
size = ticksSize,color="red") +
ggplot2::scale_x_continuous(expand = c(0, 0),limits = c(0,length(statsAdj)+1))+theme_bw()+
ggplot2::theme(panel.grid = element_blank(),
axis.text.x=element_blank(),axis.ticks.x=element_blank(),
axis.text.y=element_blank(),axis.ticks.y=element_blank())+
ggplot2::labs(x=NULL,y=NULL)+
ggplot2::theme(plot.margin = unit(c(0,5,0,5), "mm"))
dat <- data.frame(name=1:length(statsAdj),value=statsAdj)
g3 <- ggplot2::ggplot(dat,aes(name,value))+
ggplot2::geom_bar(stat="identity",fill="gray")+
ggplot2::theme_bw()+
ggplot2::theme(panel.grid = element_blank())+
ggplot2::labs(x=NULL)+
ggplot2::scale_x_continuous(expand = c(0, 0),limits=c(0,length(statsAdj)+1))+
ggplot2::theme(plot.margin = unit(c(0,5,5,5), "mm"))
p1 <- cowplot::plot_grid(plotlist = list(g1,g2,g3),nrow=3,
rel_heights = c(1,0.2,0.5),scale=1,
align ="v",labels=paste0("MITOCHONDRIAL"," pval=",pval,";NES=",nes),label_size = 8)
ggsave("result/Figure4/S5I_GSEA_B_MITOCHONDRIAL.pdf",p1,width=4,height = 4)
#------------------------------------------------------------------------------
# Step 3: Cholesterol biosynthesis pathway
#------------------------------------------------------------------------------
library(dplyr)
library(ggplot2)
## Load data and set parameters
gene_all <- data.table::fread("result/Figure4/S5H_gene_B.txt") %>%
as.data.frame()
a <- readLines("raw_data/Cholesterol.txt")
gmt <- strsplit(a, "\t")
names(gmt) <- vapply(gmt, function(y) y[1], character(1))
gmt1 <- lapply(gmt, "[", -c(1:2))
a_temp <- gene_all$logFC
names(a_temp) <- gene_all$name
Ranks_all <- sort(a_temp,decreasing =TRUE)
gseaParam = 0.5
minSize=5
ticksSize = 0.2
Ranks_all = sort(a_temp,decreasing =TRUE)
fgseaRes_all <- fgsea::fgsea(gmt1, sort(a_temp,decreasing =TRUE),minSize = minSize,nPermSimple=20000,gseaParam=0.5)
pval <- fgseaRes_all %>%
dplyr::pull(pval) %>%
round(3)
nes <- fgseaRes_all %>%
dplyr::pull(NES) %>%
round(3)
pathway <- gmt1[[1]]
stats <- Ranks_all
rnk <- rank(-stats)
ord <- order(rnk)
statsAdj <- stats[ord]
statsAdj <- sign(statsAdj) * (abs(statsAdj)^gseaParam)
statsAdj <- statsAdj/max(abs(statsAdj))
pathway <- unname(as.vector(na.omit(match(pathway, names(statsAdj)))))
pathway <- sort(pathway)
gseaRes <- fgsea::calcGseaStat(statsAdj, selectedStats = pathway,
returnAllExtremes = TRUE)
bottoms <- gseaRes$bottoms
tops <- gseaRes$tops
n <- length(statsAdj)
xs <- as.vector(rbind(pathway - 1, pathway))
ys <- as.vector(rbind(bottoms, tops))
toPlot <- data.frame(x = c(0, xs, n + 1), y = c(0, ys, 0))
diff <- (max(tops) - min(bottoms))/8
x = y = NULL
## Visualization
g1 <- ggplot2::ggplot(toPlot, aes(x = x, y = y)) +
ggplot2::geom_point(color = "green", size = 0.1) +
ggplot2::geom_hline(yintercept = 0,
colour = "gray") + geom_line(color = "green") +
ggplot2::geom_line(color = "green") + theme_bw() +
ggplot2::theme(panel.grid.minor = ggplot2::element_blank(),
axis.text.x=ggplot2::element_blank(),axis.ticks.x=ggplot2::element_blank()) +
ggplot2::labs(x = NULL, y = "enrichment score",title=)+
ggplot2::scale_x_continuous(expand = c(0, 0),limits = c(0,length(statsAdj)+1))+
ggplot2::theme(plot.margin = unit(c(5,5,0,5), "mm"))
g2 <- ggplot2::ggplot(toPlot, aes(x = x, y = y))+
ggplot2::geom_segment(data = data.frame(x = pathway),
mapping = aes(x = x,
y = -1, xend = x, yend = 1),
size = ticksSize,color="red") +
ggplot2::scale_x_continuous(expand = c(0, 0),limits = c(0,length(statsAdj)+1))+theme_bw()+
ggplot2::theme(panel.grid = element_blank(),
axis.text.x=element_blank(),axis.ticks.x=element_blank(),
axis.text.y=element_blank(),axis.ticks.y=element_blank())+
ggplot2::labs(x=NULL,y=NULL)+
ggplot2::theme(plot.margin = unit(c(0,5,0,5), "mm"))
dat <- data.frame(name=1:length(statsAdj),value=statsAdj)
g3 <- ggplot2::ggplot(dat,aes(name,value))+
ggplot2::geom_bar(stat="identity",fill="gray")+
ggplot2::theme_bw()+
ggplot2::theme(panel.grid = element_blank())+
ggplot2::labs(x=NULL)+
ggplot2::scale_x_continuous(expand = c(0, 0),limits=c(0,length(statsAdj)+1))+
ggplot2::theme(plot.margin = unit(c(0,5,5,5), "mm"))
p1 <- cowplot::plot_grid(plotlist = list(g1,g2,g3),nrow=3,
rel_heights = c(1,0.2,0.5),scale=1,
align ="v",labels=paste0("Cholesterol Biosynthesis"," pval=",pval,";NES=",nes),label_size = 8)
ggsave("result/Figure4/S5J_GSEA_B_Cholesterol.pdf",p1,width=4,height = 4)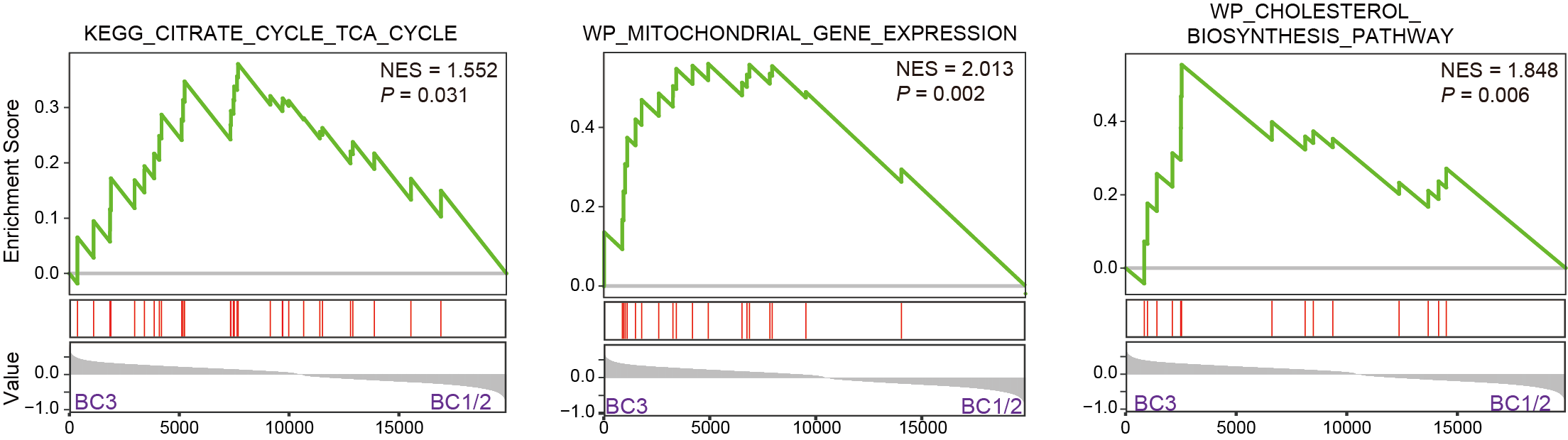
5.13 (S6 A) Clusters of T-ALL
The relative abundance of input metabolites and expression of metabolic genes in the two metabolic clusters: TC1 (N=10) and TC2 (N=14). Each column represents a sample and each row represents a metabolite (upper panel) or gene included (lower panel).
library(dplyr)
sample_info <- data.table::fread("result/Figure4/4F_cluster_T.txt") %>%
as.data.frame()
metabolite_norm = data.table::fread("result/Figure4/4F_metabolite_norm_T.txt") %>%
as.data.frame() %>%
tibble::column_to_rownames("V1")
metgene_norm <- data.table::fread("result/Figure4/4F_metgene_norm_T.txt") %>%
as.data.frame() %>%
tibble::column_to_rownames("V1")
top_annotation = HeatmapAnnotation(
Subtype=sample_info$Clusters,
col=list(Subtype=c("TC1"="#9BD53F","TC2"="#A6CEE3")),
show_annotation_name = c(T,T),
border = c(T,T) ,
simple_anno_size = unit(4, "mm"),gap = unit(1, "mm"))
col_fun = circlize::colorRamp2(c(-1, 0, 1), c("#00599F", "white", "#D01910"))
p_metabolite <- Heatmap(t(metabolite_norm),height=unit(10,"cm"),
name="Metabolite Data",
cluster_row_slices = F,clustering_method_rows = "ward.D",
clustering_method_columns = "ward.D2",
column_split = sample_info$Clusters,
col = col_fun,
top_annotation =top_annotation,
show_column_names = F,show_row_names = F,
row_names_gp = gpar(fontsize = 3),
column_names_gp = gpar(fontsize = 3))
col_fun = circlize::colorRamp2(c(-1, 0, 1), c("#00599F", "white", "#FF7F00"))
p_gene <- Heatmap(t(metgene_norm),height=unit(10,"cm"),name="Gene Data",
cluster_row_slices = F,clustering_method_rows = "ward.D",
clustering_method_columns = "ward.D2",
column_split = sample_info$Clusters,
col = col_fun,
show_column_names = F,show_row_names = F,
row_names_gp = gpar(fontsize = 3),
column_names_gp = gpar(fontsize = 3))
pdf(paste0("result/Figure4/S6A_heatmap_T.pdf"),width=8,height = 10)
p_metabolite %v% p_gene
dev.off()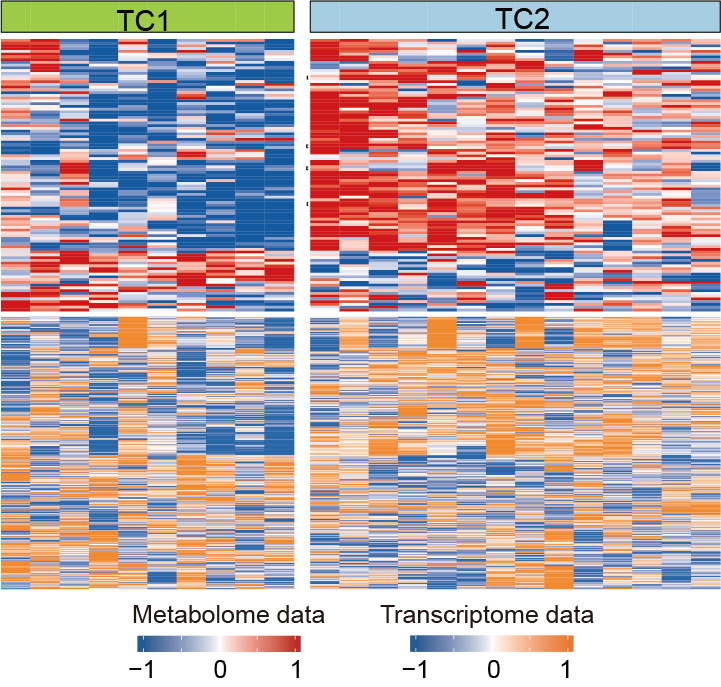
5.14 (S6 E-F) RFS and EFS of TC1/2
The associations of the two metabolic clusters with clinical RFS (E) and EFS (F) outcomes in T-ALL patients. The results suggests TC1 could be of significantly poorer prognosis than TC2.
library(dplyr)
library(survival)
library(ggplot2)
#------------------------------------------------------------------------------
# Step 1: RFS of TC1/2
#------------------------------------------------------------------------------
survival_new <- readxl::read_excel("raw_data/Meta_Files.xlsx",skip=1) %>%
as.data.frame() %>%
dplyr::select(PID,OS_stat,`OS (days)`,"EFS_stat","EFS (days)","RFS_stat","RFS (days)" ) %>%
dplyr::rename("OS"="OS (days)","EFS"="EFS (days)","RFS"="RFS (days)")
clinical_survival <- data.table::fread(paste0("result/Figure4/4F_cluster_T.txt")) %>%
as.data.frame() %>%
dplyr::select(-OS_stat,-OS,-EFS_stat,-EFS,-RFS_stat,-RFS) %>%
left_join(survival_new,by=c("Pid"="PID"))
surv_object <- Surv(time = clinical_survival$RFS/30, event = clinical_survival$RFS_stat)
fit_cluster <- survfit(surv_object ~ Clusters, data = clinical_survival)
p1 <- survminer::ggsurvplot(fit_cluster, data = clinical_survival,
title="RFS",
palette=c("#9BD53F","#A6CEE3","#EA644C"),
tables.theme = theme_void(),risk.table = TRUE,
risk.table.fontsize=6,
break.time.by = 12,
risk.table.y.text = FALSE, tables.height = 0.15,risk.table.col = "black",
pval = TRUE,pval.method = F,pval.coord=c(0.5,0.1),pval.size=6,
font.tickslab=c(14,"plain","black"),
xlab="Time (Months) ",ylab="RFS Survival")
pdf(paste0("result/Figure4/S6E_RFS_T.pdf"),onefile=FALSE)
p1
dev.off()
#------------------------------------------------------------------------------
# Step 2: EFS
#------------------------------------------------------------------------------
clinical_survival <- data.table::fread(paste0("result/Figure4/4F_cluster_T.txt")) %>%
as.data.frame()
surv_object <- Surv(time = clinical_survival$EFS/30, event = clinical_survival$EFS_stat)
fit_cluster <- survfit(surv_object ~ Clusters, data = clinical_survival)
p1 <- survminer::ggsurvplot(fit_cluster, data = clinical_survival,
title="EFS",
palette=c("#9BD53F","#A6CEE3","#EA644C"),
tables.theme = theme_void(),risk.table = TRUE,
risk.table.fontsize=6,
break.time.by = 12,
risk.table.y.text = FALSE, tables.height = 0.15,risk.table.col = "black",
pval = TRUE,pval.method = F,pval.coord=c(0.5,0.1),pval.size=6,
font.tickslab=c(14,"plain","black"),
xlab="Time (Months) ",ylab="EFS Survival")
pdf(paste0("result/Figure4/S6F_EFS_T.pdf"),onefile=FALSE)
p1
dev.off()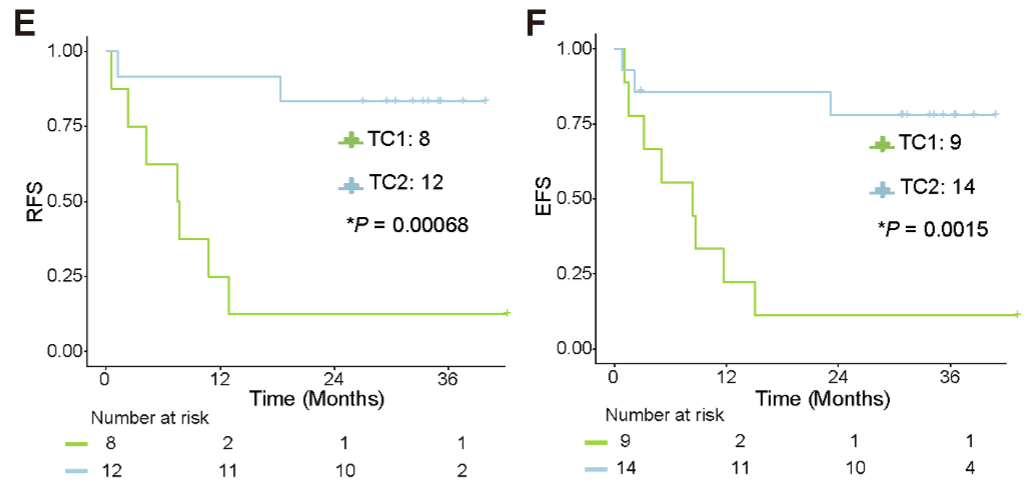
5.15 (S6 H) Multivariate Cox Analysis of TC1/2
The multivariate Cox analysis suggests TC1 may represent an unfavorable group independent of HSCT status. The WBC was stratified by 100×109/L, genetic risk was evaluated according to the NCCN guideline (V 2.2023) including RAS signaling, PTEN and NOTCH1/FBXW7, RAS/PTEN mutation and/or NOTCH1/FBXW7 wildtype were considered as having high risk, otherwise with low risk.
library(dplyr)
library(openxlsx)
library(survival)
library(survminer)
#------------------------------------------------------------------------------
# Step 1: Load data and set parameters
#------------------------------------------------------------------------------
M_label <- data.table::fread("result/Figure4/4F_cluster_T.txt") %>%
as.data.frame() %>%
dplyr::mutate(Clusters=as.factor(Clusters)) %>%
dplyr::select(Clusters,Pid)
ras <- data.table::fread("raw_data/RAS.txt") %>%
as.data.frame() %>%
dplyr::mutate(name=stringr::str_to_upper(name))
mut <- readxl::read_excel("raw_data/Sample_RNA_190_fusion_mutation_v1130.xlsx") %>%
dplyr::select(mutation,RNA_id,bianhao) %>%
tidyr::separate_rows(mutation,sep=";") %>%
dplyr::mutate(mutation=stringr::str_to_upper(mutation)) %>%
dplyr::mutate(ras=ifelse(mutation %in% ras$name,"yes","no")) %>%
dplyr::group_by(RNA_id,bianhao) %>%
dplyr::summarise(Ras1=paste(ras,collapse = ";"),mutation1=paste(mutation,collapse = ";")) %>%
dplyr::mutate(ras_new=ifelse(grepl("yes",Ras1),"yes","no"))
genetic <- read.xlsx("raw_data/Sample_RNA_190_fusion_mutation_v1130.xlsx") %>%
dplyr::select(bianhao,fusion,mutation) %>%
inner_join(mut,by="bianhao")
names(genetic)
T_gene <- genetic %>%
dplyr::select(mutation,bianhao,ras_new) %>%
dplyr::mutate(RAS_PTEN=ifelse(ras_new=="yes","MUT",
ifelse(grepl("PTEN",mutation),"MUT","WT"))) %>%
dplyr::mutate(NOTCH1_FBXW7=ifelse(grepl("NOTCH1",mutation),"MUT",
ifelse(grepl("FBXW7",mutation),"MUT", "WT"))) %>%
dplyr::mutate(risk=ifelse(RAS_PTEN=="WT"& NOTCH1_FBXW7=="MUT","Low","High")) %>%
dplyr::select(bianhao,risk) %>%
dplyr::mutate(Pid=gsub("A","RJ-",bianhao))
##### new survival #####
clinical_survival <- readxl::read_excel("raw_data/Meta_Files.xlsx",skip=1) %>%
as.data.frame() %>%
dplyr::rename("OS"="OS (days)","EFS"="EFS (days)","RFS"="RFS (days)","ifHSCT"="HSCT_stat",
"WBC"="WBC (10⁹/L)","Age"="Age (yrs)","Pid"="PID","RNA_id_raw"="RNA_ID_raw") %>%
dplyr::select(Pid,RNA_id_raw,Age,Sex,WBC,ifHSCT,OS,OS_stat,EFS,EFS_stat,RFS,RFS_stat,D28_status) %>%
dplyr::inner_join(M_label,by="Pid") %>%
dplyr::mutate(WBC_type= ifelse(WBC > 100, "High",
ifelse(WBC <= 100,"Low",WBC))) %>%
dplyr::mutate(ifHSCT=as.factor(ifHSCT)) %>%
dplyr::mutate(Age=ifelse(Age>40,"old","young")) %>%
dplyr::mutate(Age=factor(Age,levels=c("young","old"))) %>%
dplyr::mutate(Clusters=factor(Clusters)) %>%
unique() %>%
dplyr::left_join(T_gene,by="Pid") %>%
dplyr::mutate(Clusters=factor(Clusters,levels=c("TC2","TC1"))) %>%
dplyr::mutate(WBC_type=factor(WBC_type,levels=c("Low","High")))
var.names <- c("Clusters","WBC_type","D28_status","ifHSCT","risk")
#-------------------------------------------------------------------------------
# Step 2: EFS of T-ALL
#-------------------------------------------------------------------------------
f_efs <- as.formula(paste('Surv(EFS, EFS_stat)~',paste(var.names,collapse = "+")))
fit.final <- coxph(f_efs,data=clinical_survival)
p_efs <- ggforest(fit.final,main="EFS Hzard Ratio",cpositions=c(0.02,0.22,0.4),fontsize=0.8,
refLabel="reference",noDigits=2,data=clinical_survival)
#-------------------------------------------------------------------------------
# Step 3: OS of T-ALL
#-------------------------------------------------------------------------------
f_os <- as.formula(paste('Surv(OS, OS_stat)~',paste(var.names,collapse = "+")))
fit.os <- coxph(f_os,data=clinical_survival)
p_os <- ggforest(fit.os,main="OS Hzard Ratio",cpositions=c(0.02,0.22,0.4),fontsize=0.8,
refLabel="reference",noDigits=2,data=clinical_survival)
ggsave(p_efs,filename = "result/Figure4/S6H_HR_EFS_T.pdf",width=8,height=4)
ggsave(p_os,filename = "result/Figure4/S6H_HR_OS_T.pdf",width=8,height=4)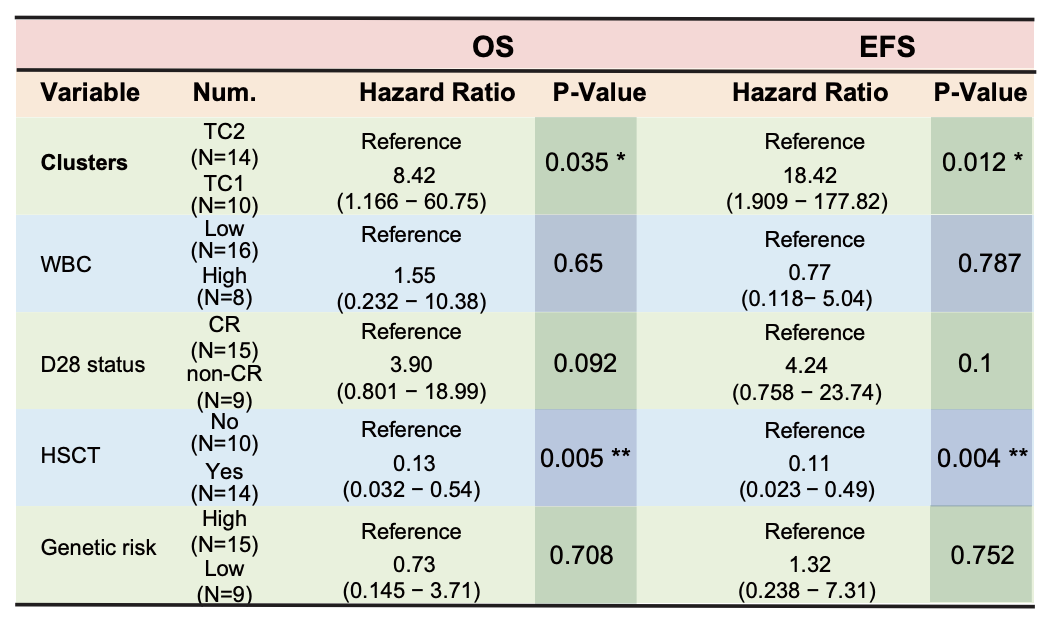
5.16 (S6 I) Waterfall plot of TC1/2
The associations of metabolic clusters with clinical variables and genetic alterations, including major mutations and fusions in T-ALL.
library(dplyr)
library(ComplexHeatmap)
#-------------------------------------------------------------------------------
# Step 1: Load data and set parameters
#-------------------------------------------------------------------------------
clinical_survival <- readxl::read_excel("raw_data/Meta_Files.xlsx",skip=1) %>%
as.data.frame() %>%
dplyr::rename("OS"="OS (days)","EFS"="EFS (days)","RFS"="RFS (days)","ifHSCT"="HSCT_stat",
"WBC"="WBC (10⁹/L)","Age"="Age (yrs)","Pid"="PID","RNA_id_raw"="RNA_ID_raw") %>%
dplyr::select(Pid,RNA_id_raw,Age,Sex,WBC,ifHSCT,OS_stat,D28_status,Subtype) %>%
dplyr::mutate(Age=ifelse(Age < 40,"< 40",">= 40")) %>%
dplyr::mutate(WBC_type= ifelse(WBC > 100, "> 100",
ifelse(WBC <= 100,"<= 100",NA))) %>%
dplyr::mutate(Outcome=ifelse(OS_stat==1,"Dead",
ifelse(OS_stat==0,"Alive",OS_stat))) %>%
dplyr::mutate(ifHSCT=ifelse(ifHSCT==0,"no",
ifelse(ifHSCT==1,"yes",ifHSCT)))
sample_info <- data.table::fread("result/Figure4/4F_cluster_T.txt") %>%
as.data.frame() %>%
dplyr::select(Clusters,Pid) %>%
dplyr::mutate(bianhao=gsub("RJ-","A",Pid)) %>%
dplyr::left_join(clinical_survival,by="Pid")
q12 <- c("#1F78B4","#E31A1C","black")
names(q12) <- unique(sample_info$Clusters)
top_annotation = HeatmapAnnotation(
Subtype=sample_info$Clusters,
Immunophenotype=sample_info$Subtype,
ifHSCT=sample_info$ifHSCT,
WBC=sample_info$WBC_type,
Outcome=sample_info$Outcome,
col=list(Subtype=c("TC1"="#9BD53F","TC2"="#A6CEE3"),
Immunophenotype=c("ETP"="#F9EE35","non_ETP"="#E7317C"),
ifHSCT=c("yes"="#CAB2D6","no"="gray"),
WBC=c("> 100"="#FDBF6F","<= 100"="gray"),
Outcome=c("Dead"="black","Alive"="gray")
),
show_annotation_name = c(T,T),
border = c(T,T,T,T,T,T) ,
simple_anno_size = unit(8, "mm"),gap = unit(1.5, "mm")
)
#-------------------------------------------------------------------------------
# Step 2: Mutations of T-ALL
#-------------------------------------------------------------------------------
mutation_T <- data.table::fread("raw_data/mutation_T_v1114_all.txt") %>%
as.data.frame() %>%
dplyr::select(V2,V17,V19) %>%
dplyr::rename("ID"="V2","Gene.refGene"="V17","ExonicFunc.refGene"="V19") %>%
dplyr::select(ID,`Gene.refGene`,`ExonicFunc.refGene`) %>%
dplyr::mutate(`ExonicFunc.refGene`=
ifelse(`ExonicFunc.refGene` %in% c("frameshift deletion","frameshift insertion","frameshift substitution"),"Frameshift",
ifelse(`ExonicFunc.refGene` %in% c("nonframeshift deletion","nonframeshift insertion","nonframeshift substitution"),"Nonframeshift",
ifelse(`ExonicFunc.refGene` %in% ".","Splice",
ifelse(`ExonicFunc.refGene` %in% "nonsynonymous SNV","Missense",
ifelse(`ExonicFunc.refGene`=="startloss","Startloss",
ifelse(`ExonicFunc.refGene`=="stopgain","Stopgain",
ifelse(`ExonicFunc.refGene`=="synonymous SNV","Silent",`ExonicFunc.refGene`)))))))) %>%
unique() %>%
dplyr::filter(ID %in% sample_info$RNA_id_raw) %>%
dplyr::mutate(`Gene.refGene` = ifelse(`Gene.refGene` %in% c("IDH1","IDH2"),"IDH1/2",`Gene.refGene`))
mutation <- rbind(mutation_T)
hit_num <- mutation %>%
dplyr::group_by(ID,`Gene.refGene`) %>%
dplyr::summarise(n=n()) %>%
dplyr::filter(n>1)
mutation <- mutation %>%
dplyr::left_join(hit_num,by=c("ID","Gene.refGene")) %>%
dplyr::mutate(`ExonicFunc.refGene`=ifelse(!is.na(n),"Multiple",`ExonicFunc.refGene`)) %>%
dplyr::select(-n) %>%
unique() %>%
dplyr::rename("RNA_id"="ID") %>%
dplyr::filter(RNA_id %in% sample_info$RNA_id_raw)
mutation_gene_filter <- mutation %>%
dplyr::select(RNA_id,Gene.refGene) %>%
unique() %>%
dplyr::group_by(`Gene.refGene`) %>%
dplyr::summarise(n=n()) %>%
dplyr::filter(n>0)
mutation_sort <- rev(names(sort(table(mutation %>% filter(`Gene.refGene` %in% mutation_gene_filter$Gene.refGene) %>% pull(Gene.refGene)))))
mutation <- mutation %>%
dplyr::filter(`Gene.refGene` %in% mutation_gene_filter$Gene.refGene) %>%
as.data.frame() %>%
dplyr::distinct(RNA_id,Gene.refGene,.keep_all = T)
sample_info_filter <- sample_info %>%
dplyr::select(bianhao,RNA_id_raw)
mutation_dcast <- reshape2::dcast(mutation,RNA_id ~ `Gene.refGene`) %>%
dplyr::right_join(sample_info_filter,by=c("RNA_id"="RNA_id_raw")) %>%
dplyr::select(-RNA_id) %>%
tibble::column_to_rownames("bianhao") %>%
dplyr::select(all_of(mutation_sort))
mutation_dcast[is.na(mutation_dcast)] <- 0
mutation_dcast[mutation_dcast=="Missense"] <- 1
mutation_dcast[mutation_dcast=="Frameshift"] <- 2
mutation_dcast[mutation_dcast=="Nonframeshift"] <- 3
mutation_dcast[mutation_dcast=="Silent"] <- 4
mutation_dcast[mutation_dcast=="Splice"] <- 5
mutation_dcast[mutation_dcast=="Stopgain"] <- 7
mutation_dcast[mutation_dcast=="Multiple"] <- 8
mutation_dcast1 <- mutation_dcast %>%
dplyr::mutate_if(is.character,as.numeric) %>%
rowwise() %>%
dplyr::mutate(NOTCH=sum(NOTCH1)) %>%
dplyr::mutate(PHF=sum(PTEN,PHF6)) %>%
dplyr::mutate(RAS=sum(JAK3,JAK1,STAT5A,MTOR,NRAS,KRAS,TCF7,TCF7)) %>%
dplyr::mutate(Transcript=sum(MED12,RUNX1,ETV6,CNOT3,MED12,GATA3,BCOR)) %>%
dplyr::mutate(Epigenetic=sum(`IDH1/2`,DNMT3A,KMT2E,SUZ12,SETD1B,SETD2,NCOR2,
EP300,KDM6A)) %>%
dplyr::mutate(Others=sum(CCND3,CTCF,BOD1L1,CCND3,SATB1,OBSCN,DNM2,CIC)) %>%
dplyr::mutate(NOTCH=ifelse(NOTCH==0,0,9)) %>%
dplyr::mutate(PHF=ifelse(PHF==0,0,9)) %>%
dplyr::mutate(RAS=ifelse(RAS==0,0,9)) %>%
dplyr::mutate(Transcript=ifelse(Transcript==0,0,9)) %>%
dplyr::mutate(Epigenetic=ifelse(Epigenetic==0,0,9)) %>%
dplyr::mutate(Others=ifelse(Others==0,0,9)) %>%
dplyr::select(NOTCH1,NOTCH,
PHF6,PTEN,PHF,
JAK3,JAK1,MTOR,NRAS,KRAS,TCF7,TCF7,STAT5A,RAS,
ETV6,RUNX1,MED12,CNOT3,MED12,GATA3,BCOR,Transcript,
DNMT3A,SUZ12,SETD1B,`IDH1/2`,KMT2E,NCOR2,EP300,KDM6A,
Epigenetic,
CTCF,CCND3,CCND3,SATB1,OBSCN,DNM2,CIC,BOD1L1,Others) %>%
as.data.frame()
rownames(mutation_dcast1) <- rownames(mutation_dcast)
mutation_dcast <- mutation_dcast1
mutation_dcast_temp <- mutation_dcast
mutation_dcast_temp[mutation_dcast_temp!=0] <- 1
num=colSums(mutation_dcast_temp)
names(mutation_dcast) <- paste0(names(mutation_dcast)," (n=",num,")")
d_mutation <- t(mutation_dcast) %>%
as.data.frame() %>%
dplyr::select(sample_info$bianhao)
mutation_annotation <- Heatmap(d_mutation,height=unit(15,"cm"),rect_gp = gpar(col = "gray", lwd = 2),
col=c("#EEEEEE","#3987CC","#DB3D3D","#663FFB","#FF7F0E","#CC66FF","#8B564C","#339933","blue"),
cluster_columns = F,cluster_rows = F,border = TRUE,column_title = NULL,
column_split =sample_info$Clusters,
show_column_names = TRUE,
top_annotation=top_annotation,
row_names_gp = gpar(fontsize = 8),column_names_gp=gpar(fontsize=6))
#-------------------------------------------------------------------------------
# Step 3: Fusions of T-ALL
#-------------------------------------------------------------------------------
fusion_T <- readxl::read_excel("raw_data/fusion_T_v0803.xlsx",skip=1) %>%
as.data.frame() %>%
dplyr::filter(RNA_id %in% sample_info$RNA_id_raw) %>%
dplyr::select(RNA_id,final_check) %>%
dplyr::left_join(sample_info,by=c("RNA_id"="RNA_id_raw")) %>%
dplyr::select(RNA_id,final_check) %>%
dplyr::filter(!is.na(final_check))
fusion <- rbind(fusion_T)
clinical_fusion_dcast <- reshape2::dcast(fusion,RNA_id ~ final_check) %>%
dplyr::right_join(sample_info_filter,by=c("RNA_id"="RNA_id_raw")) %>%
tibble::column_to_rownames("bianhao") %>%
dplyr::select(-RNA_id)
clinical_fusion_dcast[is.na(clinical_fusion_dcast)] <- 0
clinical_fusion_dcast[clinical_fusion_dcast != 0] <- 1
d_fusion <- t(clinical_fusion_dcast) %>%
as.data.frame() %>%
dplyr::select(sample_info$bianhao) %>%
tibble::rownames_to_column(var="fusion") %>%
dplyr::arrange(match(fusion,c("SET-NUP214","MLLT10 rearrangement","NUP98","STIL-TAL1",
"KMT2A rearrangement","other"))) %>%
tibble::column_to_rownames("fusion")
fusion_annotation <- Heatmap(d_fusion,rect_gp = gpar(col = "gray", lwd = 2),
col=c("#EEEEEE","#444444"),
height=unit(2,"cm"),border = TRUE,
column_split =sample_info$Clusters,
cluster_columns = F,cluster_rows = F,column_title = NULL,
row_names_gp = gpar(fontsize = 8),column_names_gp=gpar(fontsize=6))
#-------------------------------------------------------------------------------
# Step 4: Visualization
#-------------------------------------------------------------------------------
pdf(paste0("result/Figure4/S6I_oncoplot_T.pdf"),width=7,height = 10)
mutation_annotation %v% fusion_annotation
dev.off()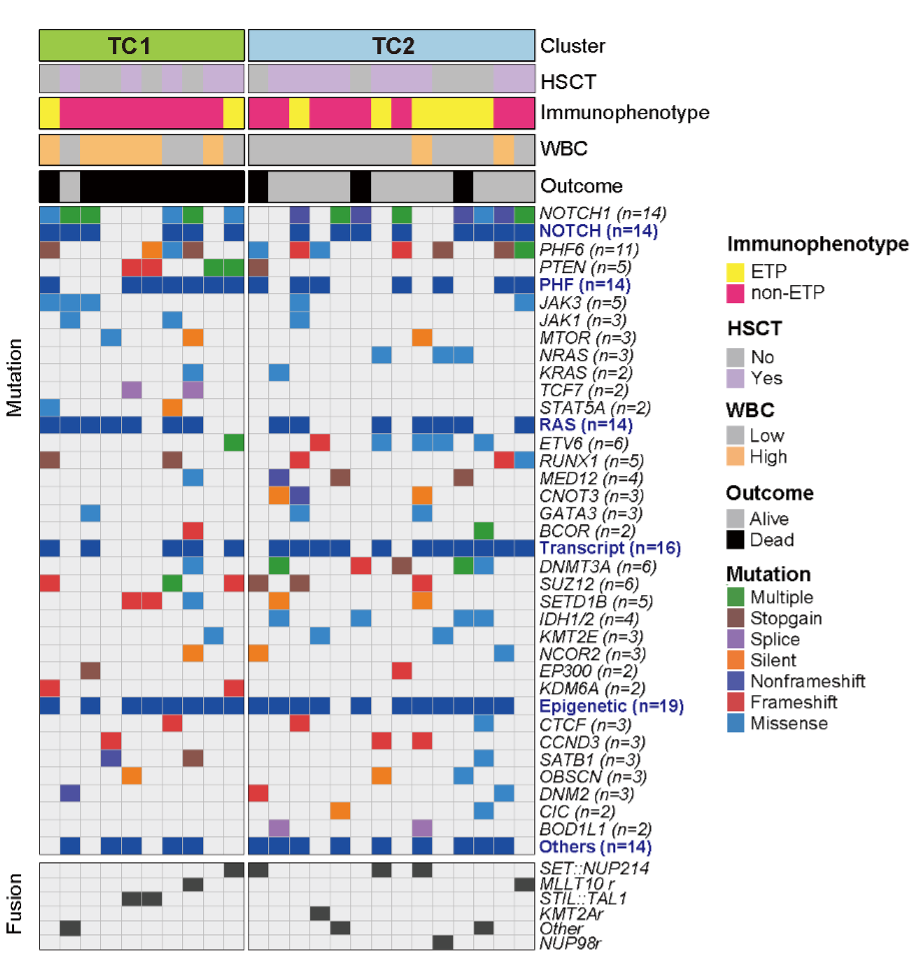
5.17 (S6 J) DA score of TC1/2
DA-score demonstrates that most of the metabolic pathways are upregulated in the relatively favorable TC2 cluster.
library(ggplot2)
library(MNet)
library(dplyr)
#------------------------------------------------------------------------------
# Step 1: DA-score of gene_T
#------------------------------------------------------------------------------
cluster <- data.table::fread("result/Figure4/4F_cluster_T.txt") %>%
as.data.frame()
dat <- data.table::fread("raw_data/cell_dat_final_reuslt_v0329.txt") %>%
dplyr::select(label,all_of(cluster$METc_BM_D0_ID)) %>%
tibble::column_to_rownames("label")
group <- cluster$Clusters
table(group)
group[which(group== "TC1")] <- "tumor"
group[which(group== "TC2")] <- "normal"
metabolite_all <- mlimma(log1p(dat),group)
write.table(metabolite_all,"result/Figure4/S6J_metabolite_T.txt",quote=F,row.names=F,sep="\t")
dd <- metabolite_all %>%
dplyr::mutate(Condition=ifelse(logFC>0 & P.Value < 0.05,"Up",
ifelse(logFC < 0 & P.Value < 0.05,"Down","normal")))
cluster <- data.table::fread("result/Figure4/4F_cluster_T.txt") %>%
as.data.frame()
dat <- readRDS("raw_data/20231113_RNA_ALL_VST_coding.rds") %>%
as.data.frame() %>%
dplyr::select(all_of(cluster$RNA_id_raw))
group <- cluster$Clusters
group[which(group== "TC1")] <- "tumor"
group[which(group== "TC2")] <- "normal"
gene_all <- mlimma(dat,group)
write.table(gene_all,"result/Figure4/S6J_gene_T.txt",quote=F,row.names=F,sep="\t")
dat <- data.table::fread("result/Figure4/S6J_gene_T.txt")
dat_increase <- dat %>%
dplyr::filter(logFC > 0) %>%
dplyr::filter(P.Value < 0.05)
dat_decrease <- dat %>%
dplyr::filter(logFC < 0) %>%
dplyr::filter(P.Value < 0.05)
DA_result <- DAscore(dat_increase$name,dat_decrease$name,dat$name,min_measured_num = 10,out="gene")
DAscore_gene_T <- DA_result$result
#------------------------------------------------------------------------------
# Step 2: DA-score of metabolite_T
#------------------------------------------------------------------------------
kegg_id <- readxl::read_excel("raw_data/cell_metabolite_info_all_v1109.xlsx") %>%
as.data.frame() %>%
dplyr::select(refmet_name,KEGG) %>%
tidyr::separate_rows(KEGG,sep=";") %>%
dplyr::filter(KEGG!="NA") %>%
dplyr::filter(KEGG != "None") %>%
as.data.frame()
dat <- data.table::fread("result/Figure4/S6J_metabolite_T.txt") %>%
dplyr::arrange(desc(logFC)) %>%
dplyr::inner_join(kegg_id,by=c("name"="refmet_name")) %>%
dplyr::distinct(KEGG,.keep_all = TRUE)
dat_increase <- dat %>%
dplyr::filter(logFC > 0) %>%
dplyr::filter(P.Value < 0.05)
dat_decrease <- dat %>%
dplyr::filter(logFC < 0) %>%
dplyr::filter(P.Value < 0.05)
DA_result <- DAscore(dat_increase$KEGG,dat_decrease$KEGG,dat$KEGG,min_measured_num = 3,out="metabolite")
write.table(DA_result$result,"result/Figure4/S6J_DAscore_metabolite_T.txt",quote=F,row.names=F,sep="\t")
#------------------------------------------------------------------------------
# Step 3: DA-score of gene+metabolite_T
#------------------------------------------------------------------------------
gene <- DAscore_gene_T %>%
as.data.frame() %>%
dplyr::filter(Measured_members_num >= 10) %>%
dplyr::filter(abs(DA_score) > 0.05)
metabolite <- data.table::fread("result/Figure4/S6J_DAscore_metabolite_T.txt") %>%
as.data.frame() %>%
dplyr::filter(Measured_members_num >= 3) %>%
dplyr::filter(DA_score != 0)
all_1 <- gene %>%
dplyr::inner_join(metabolite,by="Pathway") %>%
dplyr::filter(DA_score.x >0 & DA_score.y >0)
all_2 <- gene %>%
dplyr::inner_join(metabolite,by="Pathway") %>%
dplyr::filter(DA_score.x < 0 & DA_score.y < 0)
path <- c(all_1$Pathway,all_2$Pathway)
colp <- c("Amino acid metabolism" ="#1B9E77",
"Carbohydrate metabolism"="#D95F02",
"Glycan biosynthesis and metabolism"="#1F78B4",
"Metabolism of cofactors and vitamins"="#7570B3",
"Metabolism of terpenoids and polyketides"="#BC80BD",
"Metabolism of other amino acids"="#8DD3C7",
"Energy metabolism"="#E7298A",
"Lipid metabolism"="#66A61E",
"Nucleotide metabolism"="#E6AB02",
"Biosynthesis of other secondary metabolites"="#A6761D",
"Xenobiotics biodegradation and metabolism"="#666666")
metabolite_filter <- metabolite %>%
dplyr::filter(Pathway %in% path) %>%
dplyr::filter(Measured_members_num >= 3) %>%
dplyr::filter(abs(DA_score) > 0.05) %>%
dplyr::arrange(`Pathway Category`) %>%
dplyr::mutate(Pathway=factor(Pathway,levels = Pathway))
colp <- c("Amino acid metabolism" ="#1B9E77",
"Carbohydrate metabolism"="#D95F02",
"Glycan biosynthesis and metabolism"="#1F78B4",
"Metabolism of cofactors and vitamins"="#7570B3",
"Metabolism of terpenoids and polyketides"="#BC80BD",
"Metabolism of other amino acids"="#8DD3C7",
"Energy metabolism"="#E7298A",
"Lipid metabolism"="#66A61E",
"Nucleotide metabolism"="#E6AB02",
"Biosynthesis of other secondary metabolites"="#A6761D",
"Xenobiotics biodegradation and metabolism"="#666666")
p <- ggplot(metabolite_filter)+
geom_point(aes(x=Pathway,y=DA_score,size=log2(Measured_members_num),color=`Pathway Category`))+
geom_pointrange(aes(x=Pathway,y=DA_score,ymin=0,ymax=DA_score,color=`Pathway Category`))+
scale_color_manual(values=colp)+
coord_flip()+
xlab(NULL)+
theme_bw()
ggsave("result/Figure4/S6J_DAscore_metabolite_T_filter.pdf",p,width=8,height = 3.5)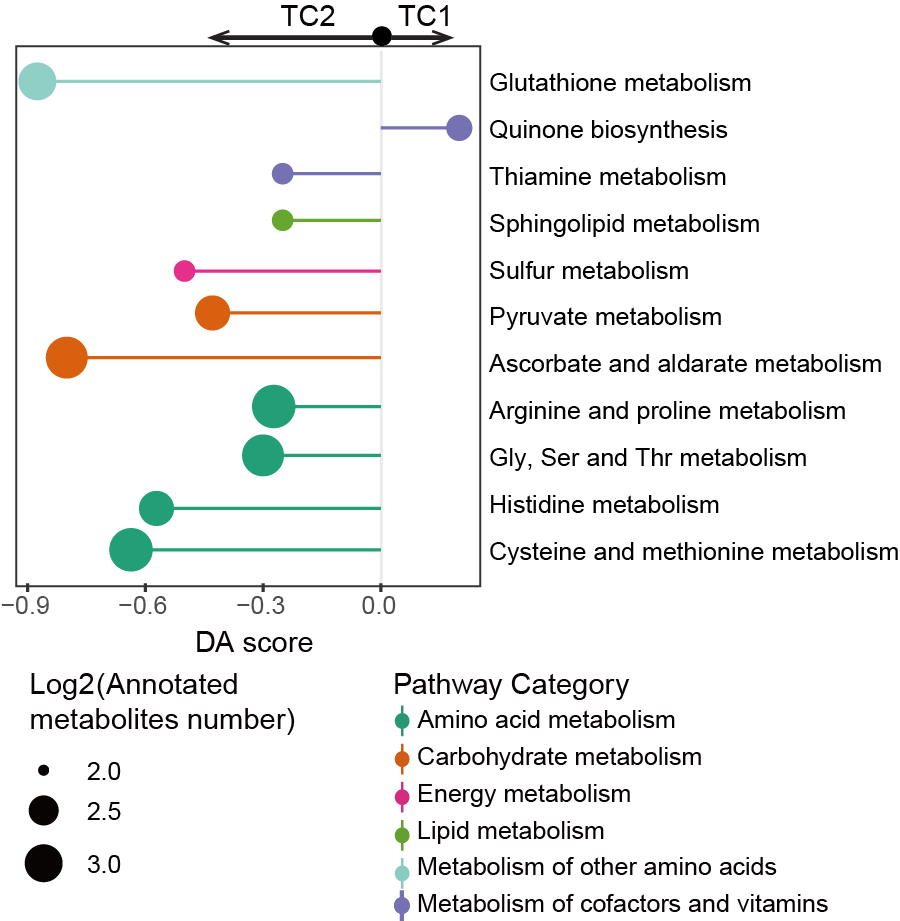
5.18 (S6 K-L) GSEA between TC1 and TC2
The GSEA reveals that the TC1 cluster, associated with poorer prognosis, exhibits suppression in OXPHOS and enrichment in histone modifications.
#------------------------------------------------------------------------------
# Step 1: Oxidative phosphorylation
#------------------------------------------------------------------------------
library(dplyr)
library(ggplot2)
## Load data
gene_all <- data.table::fread("result/Figure4/S6J_gene_T.txt") %>%
as.data.frame()
a <- readLines("raw_data/OXIDATIVE.txt")
gmt <- strsplit(a, "\t")
names(gmt) <- vapply(gmt, function(y) y[1], character(1))
gmt1 <- lapply(gmt, "[", -c(1:2))
a_temp <- gene_all$logFC
names(a_temp) <- gene_all$name
##GSEA
Ranks_all <- sort(a_temp,decreasing =TRUE)
gseaParam = 1
minSize=5
ticksSize = 0.2
Ranks_all = sort(a_temp,decreasing =TRUE)
fgseaRes_all <- fgsea::fgsea(gmt1, sort(a_temp,decreasing =TRUE),minSize = minSize,nPermSimple=20000,gseaParam=1)
pval <- fgseaRes_all %>%
dplyr::pull(pval) %>%
round(3)
nes <- fgseaRes_all %>%
dplyr::pull(NES) %>%
round(3)
pathway <- gmt1[[1]]
stats <- Ranks_all
rnk <- rank(-stats)
ord <- order(rnk)
statsAdj <- stats[ord]
statsAdj <- sign(statsAdj) * (abs(statsAdj)^gseaParam)
statsAdj <- statsAdj/max(abs(statsAdj))
pathway <- unname(as.vector(na.omit(match(pathway, names(statsAdj)))))
pathway <- sort(pathway)
gseaRes <- fgsea::calcGseaStat(statsAdj, selectedStats = pathway,
returnAllExtremes = TRUE)
bottoms <- gseaRes$bottoms
tops <- gseaRes$tops
n <- length(statsAdj)
xs <- as.vector(rbind(pathway - 1, pathway))
ys <- as.vector(rbind(bottoms, tops))
toPlot <- data.frame(x = c(0, xs, n + 1), y = c(0, ys, 0))
diff <- (max(tops) - min(bottoms))/8
x = y = NULL
g1 <- ggplot2::ggplot(toPlot, aes(x = x, y = y)) +
ggplot2::geom_point(color = "green", size = 0.1) +
ggplot2::geom_hline(yintercept = 0,
colour = "gray") + geom_line(color = "green") +
ggplot2::geom_line(color = "green") + theme_bw() +
ggplot2::theme(panel.grid.minor = ggplot2::element_blank(),
axis.text.x=ggplot2::element_blank(),axis.ticks.x=ggplot2::element_blank()) +
ggplot2::labs(x = NULL, y = "enrichment score",title=)+
ggplot2::scale_x_continuous(expand = c(0, 0),limits = c(0,length(statsAdj)+1))+
ggplot2::theme(plot.margin = unit(c(5,5,0,5), "mm"))
g2 <- ggplot2::ggplot(toPlot, aes(x = x, y = y))+
ggplot2::geom_segment(data = data.frame(x = pathway),
mapping = aes(x = x,
y = -1, xend = x, yend = 1),
size = ticksSize,color="red") +
ggplot2::scale_x_continuous(expand = c(0, 0),limits = c(0,length(statsAdj)+1))+theme_bw()+
ggplot2::theme(panel.grid = element_blank(),
axis.text.x=element_blank(),axis.ticks.x=element_blank(),
axis.text.y=element_blank(),axis.ticks.y=element_blank())+
ggplot2::labs(x=NULL,y=NULL)+
ggplot2::theme(plot.margin = unit(c(0,5,0,5), "mm"))
dat <- data.frame(name=1:length(statsAdj),value=statsAdj)
g3 <- ggplot2::ggplot(dat,aes(name,value))+
ggplot2::geom_bar(stat="identity",fill="gray")+
ggplot2::theme_bw()+
ggplot2::theme(panel.grid = element_blank())+
ggplot2::labs(x=NULL)+
ggplot2::scale_x_continuous(expand = c(0, 0),limits=c(0,length(statsAdj)+1))+
ggplot2::theme(plot.margin = unit(c(0,5,5,5), "mm"))
p1 <- cowplot::plot_grid(plotlist = list(g1,g2,g3),nrow=3,
rel_heights = c(1,0.2,0.5),scale=1,
align ="v",labels=paste0("OXIDATIVE_PHOSPHORYLATION"," pval=",pval,";NES=",nes),label_size = 8)
ggsave("result/Figure4/S6K_GSEA_T_OXIDATIVE.pdf",p1,width=5,height = 5)
#------------------------------------------------------------------------------
# Step 2: Histone modifications
#------------------------------------------------------------------------------
## Load data
gene_all <- data.table::fread("result/Figure4/S6J_gene_T.txt") %>%
as.data.frame()
a <- readLines("raw_data/histone.txt")
gmt <- strsplit(a, "\t")
names(gmt) <- vapply(gmt, function(y) y[1], character(1))
gmt1 <- lapply(gmt, "[", -c(1:2))
a_temp <- gene_all$logFC
names(a_temp) <- gene_all$name
##GSEA
Ranks_all <- sort(a_temp,decreasing =TRUE)
gseaParam = 1
minSize=5
ticksSize = 0.2
Ranks_all = sort(a_temp,decreasing =TRUE)
fgseaRes_all <- fgsea::fgsea(gmt1, sort(a_temp,decreasing =TRUE),minSize = minSize,nPermSimple=20000,gseaParam=0.5)
pval <- fgseaRes_all %>%
dplyr::pull(pval) %>%
round(3)
nes <- fgseaRes_all %>%
dplyr::pull(NES) %>%
round(3)
pathway <- gmt1[[1]]
stats <- Ranks_all
rnk <- rank(-stats)
ord <- order(rnk)
statsAdj <- stats[ord]
statsAdj <- sign(statsAdj) * (abs(statsAdj)^gseaParam)
statsAdj <- statsAdj/max(abs(statsAdj))
pathway <- unname(as.vector(na.omit(match(pathway, names(statsAdj)))))
pathway <- sort(pathway)
gseaRes <- fgsea::calcGseaStat(statsAdj, selectedStats = pathway,
returnAllExtremes = TRUE)
bottoms <- gseaRes$bottoms
tops <- gseaRes$tops
n <- length(statsAdj)
xs <- as.vector(rbind(pathway - 1, pathway))
ys <- as.vector(rbind(bottoms, tops))
toPlot <- data.frame(x = c(0, xs, n + 1), y = c(0, ys, 0))
diff <- (max(tops) - min(bottoms))/8
x = y = NULL
g1 <- ggplot2::ggplot(toPlot, aes(x = x, y = y)) +
ggplot2::geom_point(color = "green", size = 0.1) +
ggplot2::geom_hline(yintercept = 0,
colour = "gray") + geom_line(color = "green") +
ggplot2::geom_line(color = "green") + theme_bw() +
ggplot2::theme(panel.grid.minor = ggplot2::element_blank(),
axis.text.x=ggplot2::element_blank(),axis.ticks.x=ggplot2::element_blank()) +
ggplot2::labs(x = NULL, y = "enrichment score",title=)+
ggplot2::scale_x_continuous(expand = c(0, 0),limits = c(0,length(statsAdj)+1))+
ggplot2::theme(plot.margin = unit(c(5,5,0,5), "mm"))
g2 <- ggplot2::ggplot(toPlot, aes(x = x, y = y))+
ggplot2::geom_segment(data = data.frame(x = pathway),
mapping = aes(x = x,
y = -1, xend = x, yend = 1),
size = ticksSize,color="red") +
ggplot2::scale_x_continuous(expand = c(0, 0),limits = c(0,length(statsAdj)+1))+theme_bw()+
ggplot2::theme(panel.grid = element_blank(),
axis.text.x=element_blank(),axis.ticks.x=element_blank(),
axis.text.y=element_blank(),axis.ticks.y=element_blank())+
ggplot2::labs(x=NULL,y=NULL)+
ggplot2::theme(plot.margin = unit(c(0,5,0,5), "mm"))
dat <- data.frame(name=1:length(statsAdj),value=statsAdj)
g3 <- ggplot2::ggplot(dat,aes(name,value))+
ggplot2::geom_bar(stat="identity",fill="gray")+
ggplot2::theme_bw()+
ggplot2::theme(panel.grid = element_blank())+
ggplot2::labs(x=NULL)+
ggplot2::scale_x_continuous(expand = c(0, 0),limits=c(0,length(statsAdj)+1))+
ggplot2::theme(plot.margin = unit(c(0,5,5,5), "mm"))
p1 <- cowplot::plot_grid(plotlist = list(g1,g2,g3),nrow=3,
rel_heights = c(1,0.2,0.5),scale=1,
align ="v",labels=paste0("histone"," pval=",pval,";NES=",nes),label_size = 8)
ggsave("result/Figure4/S6L_GSEA_T_histone.pdf",p1,width=4,height = 4)